Type II Waardenburg Syndrome: Case Report
- 1. Resident, Hospital Otorrinos de Feira de Santana, Brazil
- 2. Hospital Otorrinos de Feira de Santana, Brazil
ABSTRACT
Introduction: Waardenburg syndrome (WS) was first described by P.J. Waardenburg. The same author estimates such syndrome is responsible for 1,43% of all congenital deafness. WS presents a variable spectrum as four clinical presentations. The diagnosis is clinical using specific criteria established in 1992 by Farrer et al. The prognosis depends on the presence of comorbidities such as delay in neuropsychomotor behavior and adequate educational intervention.
Case report: JPCS, male, 2 years old, grandmother refers deafness. Physical exam: Face inspection: hypo pigmentation of right iris, normal pigmentation of left iris. Otoscopy: normal.
Additional testing: Otoacoustic Emissions: absent bilaterally; BERA: wave I absent; Ear CT: semicircular canals dysplasia. The patient was forwarded to a hearing rehabilitation center.
Discussion: The patient presents the following major criteria for diagnosis: neurological congenial deafness, heterochromic iris, first degree relative affected. Assertive and precocious diagnosis allows audiological intervention with cochlear implants and excellent prognosis with audiological rehabilitation.
Final comments: Waardenburg Syndrome is a rare disease and with great importance particularly in pediatric population because precocious intervention has a great impact in the patient’s life quality.
KEYWORDS
• Deafness
• Congenital abnormalities
• Waardenburg syndrome
CITATION
e Santos ECC, Bem Mendonça NMC, Leite MF, da Cunha Moura MP, de Menezes Santos Torres S, de Cerqueira Almeida WL, et al. (2016) Type II Waardenburg Syndrome: Case Report. Ann Otolaryngol Rhinol 3(6): 1113.
INTRODUCTION
Waardenburg syndrome (WS) was first described by P.J.Waardenburg, ophthalmologist and Dutch geneticist in 1951. It is a genetic disease with autosomal dominant heritage patter, with incomplete penetrance and variable phenotypes. The same author estimates the incidence of this syndrome to be approximately 1.43% of patients with congenital deafness and 1: 42,000 in the general population [1]. Studies have revealed incidences ranging from 0.9% to 2.8% of congenitally deaf population [2-4].
It’s most frequent clinical signs are sensorineural hearing loss, affecting about 60% of the patients; heterochromia of the iris; hypoplastic blue eyes; white streak; premature gray hair; leucoderma; high nasal root and hyperplasia of the medial portion of the eyebrows (synophrys) [5]. WS has four clinical presentations. Type I patients have skin fold extending from the base of the nose to the end of medial eyebrow region (epicanthus), increasing the distance of the internal medial corners of the eyes (canthorum dystopia), iris isocromia with bright blue color or heterochromia of iris, white hair streak (poliosis) that can appear at any age, confluent eyebrows (sinophrys) and changes in skin pigmentation. Type II differs from type I by not showing canthorum dystopia. Type III, also known as Klein Waardenburg Syndrome, has in addition to type I characteristics, microcephaly, malformation of the upper limbs and mental disabilities. Type IV, also known as Waardenburg-Shah Syndrome presents Hirschsprung’s disease in addition to Type II manifestations, characterized by the absence of ganglion cells of Auerbach plexus and Meissner plexus [6].
Diagnostic criteria for WS were established by Farrer et al. in 1992. Major criteria are: congenital sensorineural hearing loss, hair hypopigmentation, abnormal pigmentation of the iris (complete or partial heterochromia), telecanthus, first-degree relatives affected. Minor criteria are: gray hair before the of age 30, synophrys, skin hypopigmentation, high nasal root or large, hypoplastic nose wing. The diagnosis is given by the presence of two major criteria or one major and two minor.
In current medical literature six genes are associated with WS: PAX3 (encoding the paired box 3 transcription factor), MITF (microphthalmia-associated transcription factor), EDN3 (endothelin 3), EDNRB (endothelin receptor type B), SOX10 (encoding the Sry bOX10 transcription factor) and SNAI2 (snail homolog 2). PAX3 alterations can be found in malignant tumors (such as melanomas and neuroblastomas) and in WS. It is involved in the development of the central nervous system, skeletal muscle, cardiac tissue, melanocytes and enteric ganglia. Around 70 different mutation points have been identified with very few recurrences reported. The MITF gene is known as the key to melanocyte development. Mutations in this gene occur in about 15% of WS Type II patients. The endothelins, represented by EDN3 and EDNRB, have an important role in the development of neural crest cells. Over fifteen endothelin mutations have been described in WS patients. SOX10 is involved in the early development of the neural crest; it expresses proteins linked to cell fate determination and cell linage. It also has shown the capacity to regulate PAX3 and MITF genes. These functions are crucial for cell differentiation in glial cells and oligodendrocytes. It is the main gene involved in type IV WS. SNAI2 has an essential role in germinative cells, melanocytic and hematopoietic stem cells. This gene has minor involvement in type II WS [7].
Auditory involvement is not always clear in WS, some studies show anatomical changes in computed tomography such as cochlear hypoplasia or aplasia, posterior semicircular canal aplasia, abnormal vestibule and absence of oval window [8]. The hearing prognosis depends on the cochlear morphology as well as the presence of comorbidities such as mental developmental delay and appropriate educational intervention [9].
CASE PRESENTATION
JPCS, 2 years old, male, born and raised in Feira de Santana – BA attended the ENT visit accompanied by his grandmother who mentioned pre lingual hearing loss and delayed speech development. She denied perceived visual impairment. The patient was born to term delivery, weighing 2700 grams, denies perinatal diseases. Mother attended 06 prenatal consultations uneventfully. As family history his grandmother referred auditory impairment in the patient’s mother. Denied maternal and paternal consanguinity.
On physical examination: good general condition, ruddy, hydrated, anicteric, acyanotic, afebrile, well nourished, active, not reactive, not responsive to verbal commands. Face inspection: hypo pigmented right iris (light blue-colored iris), no canthorum dystopia, no sinophrys, no skin pigmentation changes (Figure 1).
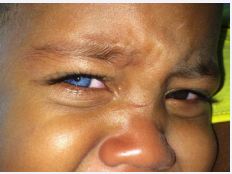
Figure 1: Heterochromia of the iris.
Extremities: no deformities of the arms or leg. Otoscopy: pervious external auditory meatus, tympanic membrane with no perforations, translucent, Oroscopy: Tonsils grade III. Rhinoscopy: eutrophic inferior turbinates, septum centered, normal colored mucosa. Trunk inspection: coffee- milk birthmark in the abdomen.
Otoacoustic emissions (OAE), Brainstem Evoked Response Audiometry (BERA) and Ear Computed Tomography (CT) were requested with the following results: OAE: absent bilaterally; BERA: wave I absent, not possible to detect the presence of retro cochlear alterations. Presence of waves III and V. Electrophysiological threshold compatible with acoustic thresholds around 75 db in specific frequencies of this examination (around 4 kHz) (Figures 2),
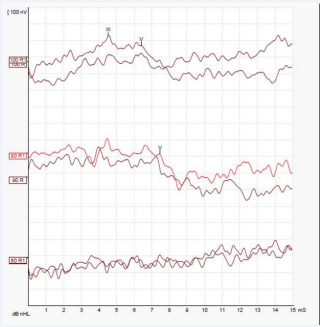
Figure 2: BERA – Right Ear
(Figures 3),
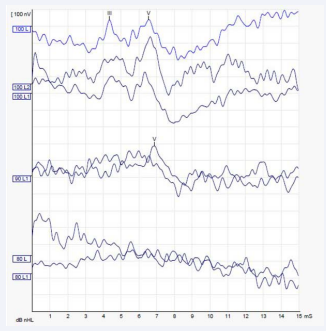
Figure 3: BERA – Left Ear
Ear CT: dysplasia of the semicircular canals and the vestibule bilaterally, amorphous content in the external auditory canal (Figure 4).
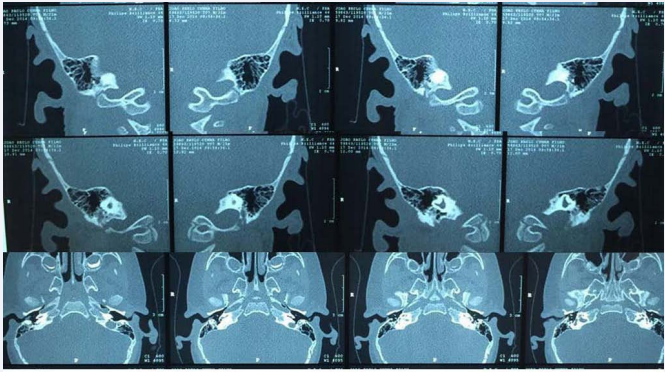
Figure 4: Ear CT: dysplasia of the semicircular canals and the vestibule bilaterally.
The patient was then referred to the cochlear implant team, which will evaluate cochlear permeability through magnetic resonance imaging.
DISCUSSION
The etiology of WS, in this particular case, is an autosomal dominant genetic anomaly transmitted by the patient’s mother. In current literature it is estimated that approximately 25% of cases are derived from new mutations consisting in sporadic cases.
The patient in question has the following major criteria: congenital sensorineural hearing loss, iris heterochromia and affected first-degree relative, being established clear clinical diagnosis of 03 major criteria. Among its manifestations, complete heterochromia of the iris is of rare penetrance.
The treatment approach should be multidisciplinary involving ophthalmology, Otorhinolaryngology and genetic counseling. Early diagnosis allows efficient audiological approach improving quality of life. Cullen et al. demonstrated effective results in the treatment of WS with cochlear implants, as well as excellent prognosis with proper hearing rehabilitation [9]. Waardenburg Syndrome is a rare disease of great importance, particularly in the pediatric population; early intervention has major impact in the patient’s life quality. Genetic counseling and multidisciplinary patient approach is crucial.









































































































































