Dementia is an Intersectional Neuropathology: A Review
- 1. Department of Molecular and Cellular Biology, Baylor College of Medicine, USA
ABSTRACT
Dementia is an increasing health problem in an aging population with over 10 million patients with the diagnosis in the US. In the several disease variations characterized by dementia there are common clinical and histological findings, suggesting shared underlying neuronal deficiencies and clinical outcome. Observations support the notion that any of several mechanisms may cause a loss of neuron function, progressing to dementia. In this review we summarize evidence leading to the conclusion that defects in separate neuronal processes are additive, leading to the clinical manifestation of dementia as a result of cumultive defects in discrete cellular functions. Therefore we conclude that the manifstations of dementia are the result of an intersection of defects
KEYWORDS
Dementia, Alzheimer’s, Parkinson’s, Huntington’s, Proteostasis
CITATION
Moses RE, O’Malley BW (2020) Dementia is an Intersectional Neuropathology: A Review. Ann Clin Pathol 7(1): 1152.
INTRODUCTION
Neurodegenerative disease has multiple variations, including frontotemporal dementia, microvascular atherosclerosis, and Huntington’s (HD) disease. The most common are Alzheimer’s (AD), and Parkinson’s (PD): more than six million Americans live with Alzheimer’s disease, and at least one million Americans live with Parkinson’s disease. Since age is the primary risk factor, dementia resulting from these diseases is an increasing problem as the population of the country ages. Dementia affects 7-10% of persons over age 65 with a higher risk in more developed countries due to longer life span [1-3].
Dementia is a syndrome with multiple aspects, as indexed by the Diagnostic and Statistical Manual of Mental Disorders-V: activity changes such as purposeless motion, loss of ability to perform daily living tasks, inattention, wandering, mood changes, and thought disorders. It is characterized by a decrease in cognition that leads to a loss of prior performance standards in social, occupational or domestic functioning [2,4]. Alzheimer designated patients with dementia and findings of neurofibrillary tangles and neuritic plaques (NP) in the central nervous system (CNS), often are thought to be on a vascular basis. AD was considered a ‘pre-senile’ dementia for decades but by the 1970’s AD and dementia occurring in old age came to be viewed as a single process [5-7] because of common manifestations and CNS findings. Presently 60-80% of diagnoses for dementia are defined as AD. Because the findings in AD and senile dementia are the same histologically and behaviorally, we will treat AD in this discussion as a valid model for dementia seen with aging.
Although the major predisposing risk factor for dementia is age [8,9], it appears that some of the risk factors for dementia, for example hypertension, affect onset by age 65, but may be of diminished effect after age 85 [10,11]. Intriguingly, tissues from ‘normal’ aged brains may show histological findings associated with AD, and not all clinical diagnoses of AD show the typical tissue findings of neurofibrillary tangles and NP [12]. Improved imaging methods hold the promise of better pre-symptomatic detection [4,12-16].
Pre-clinical research has been hampered by the lack of suitable animal models for dementia and the working field has not defined the specific underlying causes of these diseases. Why is this? Perhaps because of excessive focus on the late-stage disease pathology, and not on discovery of the triggering etiologies. The field has become ‘entangled in CNS tangles’, composed of proteins such as β-Amyloid, α-synuclein, tau bodies, and Huntingtin in the different diseases. These protein deposits are evident in the brain of advanced neurodegenerative disease patients and serve as pathologic and diagnostic markers for AD or PD, for example. Such marker proteins may be seen as endpoint consequences of disease etiology. They do, however, contribute to the severity of the symptoms because deposits of amyloid, synuclein, and tau share a 3-D structure that has been reported to impair entry into the proteasome and its function [17]. These aggregate proteins also interfere with synapse connections and functions when secreted into the brain extracellular space. Such findings emphasize the importance of efficient cellular proteolysis for brain function. Recent research indicates that these protein structures can seed in a prion-like manner and also spread via exosomes, trans-synaptically and via axons from affected cells to normal [18-24]. Attempts to decrease late-stage accumulation of β−Amyloid by use of antibody agents have been unsuccessful in slowing the pace, onset, or progression of AD [25,26].
There are several schools of thought regarding the primary causes of dementia. The categories include metabolic, familial, vascular, inflammatory, hypertension, blood brain barrier, lymphatic and toxic [4,15,27-31]. Our hypothesis is that there are several mechanisms for loss of neuronal function and when the sum effect on the aging cell creates sufficient impairment of neuronal function, and enough cells are affected, then dementia becomes clinically apparent. Based on that hypothesis, we think that dementia is a neuropathy caused by accumulation of defects in any of several interactive processes, defining it as a multifactorial, intersectional neuropathy. For dementia to become apparent it is not necessary that neurons die; only that sufficient numbers become defective in function to a great enough degree. Here we will address several major hypotheses for neurodegeneration leading to dementia with particular attention on protein homeostasis.
GENETIC/FAMILIAL
A minority of cases of AD are of a dominant heritable basis and early onset, for example patients with mutations in Amyloid Precursor Protein (APP) [32,33]. Patients with familial AD (FAD) have a mutation in one of three genes known to act in the development of AD: PS1, PS2 and AAP. Another predisposing mutation is in the ApoE4 protein [34,35]. Carriers of APOE4 are more likely to develop AD; although APOE4 is present in only 15% of the population, it is present in a majority of late onset AD; a single allele increase risk for AD by 2-4-fold and two alleles by 8-12-fold [36].
INFLAMMATION
Some observations favor the concept that neuroinflammation is the initial step in loss of central nervous system function and dementia [37,38]. There is evidence supporting the concept of the inflammasome as a driving etiology for neurodegeneration [39,40]. Glial cells and astrocytes become inflamed and secrete inflammatory cytokines leading to loss of neuronal function. Inflammation could sponsor more β-Amyloid deposition, promoting neurodegeneration. Improved imaging techniques may allow inflammation to be detected and tracked [37]. It seems reasonable to favor the inflammasome as one of the driving factors, but not the sole initiating one, for dementia.
ATHERSCLEROSIS AND MICROVASCULAR EVENTS
The original obervations of AD favored the idea that loss of blood flow and mini-strokes led to loss of neuronal function and hence to dementia. Atherosclersis-based microvascular defects are a major basis for loss of neuronal capability [13,41,42] and there is robust evidence for microvascular defects in AD. It has been suggested that neuritic plaques cause a decrease in blood flow, and thus amyloid plaques and microvasuclar strokes, along with decreased cerebral blood flow due to atherosclerosis, would act synergistically to hamper neuronal function.
METABOLIC
There are several long-recognized metabolic deficiencies associated with dementia. For example, Korsakoff syndrome secondary to chronic alcoholism (deficiency of B1), and pellagra (deficiency of B3). Low NAD+ may be one final common step of these processes [1, 43-46]. With age, intracellular NAD+ levels decline and mitochondrial function is known to be decreased, as in later stages of dementia. A possible interaction between NAD+ and reactive oxygen species (ROS) in the mitochondrial has been speculated to affect AD [45]. Overall we favor the idea that the energy state of the neuron is a central factor in neuronal health and that a decrease in NAD+ with age [43,46] favors development of dementia.
DECREASED PROTEOSTASIS
With age, people gradually experience accumulating dysfunction of their biological processes. We become less able to do physical (muscular) work, to react quickly and coordinately, to calculate, to recall names rapidly, to sleep soundly and to accommodate stress.
Protein turnover is a regulated cellular process and nowhere more so than in the brain. Both cytoplasmic and mitochondrial proteins undergo a programmed destruction by autophagy and mitophagy, producing components for re-use by the cell, conserving energy. The regulated maintenance of protein homeostasis is referred to as proteostasis. A major factor in the ordered and accurate turnover is the 26S proteasome [47]. We have described an alternate proteasome pathway whose deficiency may play a significant role in plaque accumulation, the REGγ pathway that is ATP- and Ub- independent [48]. The loss of programmed protein turnover may promote the appearance of β-Amyloid plaques; therefore, we choose to place plaque formation and synaptic decay under this rubric [21, 49-52].
Aberrant or partially degraded proteins accumulating due to loss of proteostasis elicit a cellular endoplasmic reticulum (ER) stress response, the unfolded protein response (UPR). The ER UPR depends on several pathways [53] with the X Box binding protein (XBP1) acting as a master regulator of the stress response. This pathway is essential for maintenance of homeostasis for protein degradation, and hence mitochondrial mitophagy. A decrease of the ER stress response, normal with increasing age, plus the secondary lack of proteostasis have been suggested to be drivers of AD [54,55]. The UPR leads to an inflammatory cascade. This supports the concept that accumulation of normal or aberrant proteins, as is common in neurodegenerative disease, is a late marker of disease and not the primary cause. We conclude that evidence favors defective proteostasis as an initiating and main driving element for neurodegeneration.
CONCLUSIONS AND PREDICTIONS
The evidence leads us to propose that clinical dementia results from cumulative defects in any of several pathways that lead to a clinical presentation when the sum crosses a functional threshold (See Figure 1).
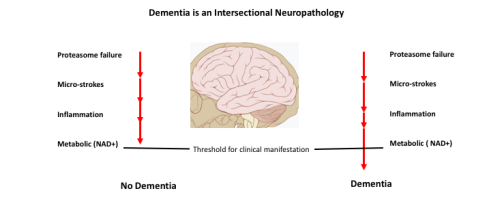
Figure 1 Loss of neuronal function leading to dementia is based on accumulation of defects in any of several systems. Once a threshold of dysfunction is crossed, and enough cells are affected, dementia is established.
Our conclusions support several predictions.
• Our multifactorial model of dementia yields targets for early diagnosis and potential therapeutic approaches that can be aimed at preventing initiation of the degeneration.
• The subdivision of independent but cumulative causes of dementia, coupled with improved pre-clinical diagnostics, can provide improved metrics to monitor evolving dementia.
• Cell death is not an absolute requirement for dementia.
• New therapies that rejuvenate the cell are likely to be drugs that return the cell to an early and more embryonic/ developmental transcriptional state.
REFERENCES
- Hou Y, Dan. X Babbar Wei. Y, Hasselbalch. SG, Croteau DL, Bohr VA. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019; 15: 565-581.
- Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and Management of Dementia: Review. JAMA. 2019; 322: 1589-1599.
- Ryu JC, Zimmer ER, Rosa-Neto P, Yoon SO. Consequences of Metabolic Disruption in Alzheimer's Disease Pathology. Neurotherapeutics. 2019; 16: 600-610.
- Gale SA, Acar D, Daffner KR. Dementia. Am J Med. 2018; 131: 1161-1169.
- Drachman DA. If we live long enough, will we all be demented?, Neurology. 1994; 44 :1563-1565.
- Haase GR. Diseases presenting as dementia, Contemp Neurol Ser. 1997; 15: 27-67.
- Samorajski T. How the human brain responds to aging. J Am Geriatr Soc. 1976; 24: 4-11.
- Ferrari C, Lombard G, Polito C, Lucidi G, Bagnoli S, Piaceri I, et al. Alzheimer's Disease Progression: Factors Influencing Cognitive Decline. J Alzheimers Dis. 2018; 61: 785-791.
- Boccardi V, Comanducci C, Baroni M, Mecocci P. Of Energy and Entropy: The Ineluctable Impact of Aging in Old Age Dementia, Int J Mol Sci .2017; 18: 2672.
- Gardner RC, Valcour V, Yaffe K. Dementia in the oldest old: a multi-factorial and growing public health issue. Alzheimers Res Ther. 2013; 5: 27.
- Euser SM, van Bemmel T, Schram MT, Gussekloo J, Hofman A, Westendorp RG, et al. The effect of age on the association between blood pressure and cognitive function later in life. J Am Geriatr Soc. 2009; 57: 1232-1237.
- Blamire AM. MR approaches in neurodegenerative disorders, Prog Nucl Magn Reson Spectrosc. 2018; 108: 1-16.
- Liesz A. The vascular side of Alzheimer's disease. Science. 2019; 365: 223-224.
- Salloway S. Improving Evaluation of Patients With Cognitive Impairment With Amyloid Positron Emission Tomography. JAMA Neurol. 2018; 75: 1045-1046.
- McLachlan E, Rai S, Al-Shihabi A, Huntley J, Burgess N, Howard R, Reeves S. Neuroimaging correlates of false memory in 'Alzheimer's disease: A preliminary systematic review. Psychiatry Res Neuroimaging. 2019; 296: 111021.
- Frantellizzi V, Morreale M, Pontico M, Francia A, Drudi F.M, Farcomeni A. Liberatore M, (99m) Tc-HMPAO brain SPECT in the monitoring of cerebral vasculitis therapy. Rev Esp Med Nucl Imagen Mol. 2018; 37: 211-217.
- Hong L, Huang HC, Jiang ZF. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer's disease, Neurol Res. 2014; 36: 276-282.
- Surmeier DJ, Obeso JA, Halliday GM. Parkinson's Disease Is Not Simply a Prion Disorder. J Neurosci. 2017; 37: 9799-9807.
- Chen XQ, Mobley WC. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Abeta and Tau Species. Front Neurosci. 2019; 13: 659.
- Ruiz-Riquelme A, Lau HH, Stuart E, Goczi AN, Wang Z, Schmitt-Ulms G, et al. Prion-like propagation of beta-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol Commun. 2018; 6: 26.
- Sarnataro D. Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases, Int J Mol Sci. 2018; 19: 3081.
- Vaquer-Alicea J, Diamond MI. Propagation of Protein Aggregation in Neurodegenerative Diseases. Annu Rev Biochem. 2019; 88: 785-810.
- Maxan A, Cicchetti F. Tau: A Common Denominator and Therapeutic Target for Neurodegenerative Disorders. J Exp Neurosc. 2018; 12: 1179069518772380.
- Masnata M, Cicchetti F. The Evidence for the Spread and Seeding Capacities of the Mutant Huntingtin Protein in in Vitro Systems and Their Therapeutic Implications, Front Neurosci. 2017; 11: 647.
- Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, et al. A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018; 90: e1889-e1897.
- Golde TE, DeKosky ST, Galasko D. Alzheimer's disease: The right drug, the right time. Science. 2018; 362: 1250-1251.
- Hachinski V, Einhaupl K, Ganten D, Alladi S, Brayne C, Stephan BCM, et al. Preventing dementia by preventing stroke: The Berlin Manifesto. Alzheimers Dement. 2019; 15: 961-984.
- Skaper SD, Facci L, Zusso M, Giusti P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front Cell Neurosci. 2018; 12: 72.
- Skaper SD. Impact of Inflammation on the Blood-Neural Barrier and Blood-Nerve Interface: From Review to Therapeutic Preview. Int Rev Neurobiol. 2017; 137: 29-45.
- Mogi M. Could Management of Blood Pressure Prevent Dementia in the elderly?, Clin Hypertens. 2019; 25: 27.
- [Ueo M, Chiba Y, Murakami R, Matsumoto K, Fujihara R, Uemura N, Yanase K, Kamada M. Disturbance of Intracerebral Fluid Clearance and Blood-Brain Barrier in Vascular Cognitive Impairment. Int J Mol Sci. 2019; 20: 2600.
- Drachman DA. Aging of the brain, entropy, and Alzheimer disease. Neurology. 2006; 67: 1340-1352.
- Rosenberg RN, Lambracht-Washington D, Xia G. Yu, W. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016; 7: 867-874.
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kurkull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997; 27: 1349-1356.
- Lane-Donovan C, Herz J. ApoE Receptors, and the Synapse in Alzheimer's Disease. Trends Endocrinol Metab. 2017; 28: 273-284.
- Belloy ME, Napolioni V, Greicius MD. A Quarter Century of APOE and Alzheimer's Disease: Progress to Date and the Path Forward. Neuron. 2019; 101: 820-838.
- Schain M, Kreisl WC. Neuroinflammation in Neurodegenerative Disorders-a Review. Curr Neurol Neurosci Rep. 2017; 17: 25.
- uzman-Martinez LG, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos-Escobar N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front Pharmacol. 2019; 10: 1008.
- Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. 2019; 11: e10248.
- Sun F, Deng Y, Han X, Liu Q, Zhang P, Manzoor R, Ma H. A secret that underlies Parkinson's disease: The damaging cycle. Neurochem Int. 2019; 129: 104484.
- Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 2019; 18: 684-696.
- Hansra GK, Popov G, Banaczek PO, Vogiatzis M, Jegathees T, Goldbury CS,et al. The neuritic plaque in Alzheimer's disease: perivascular degeneration of neuronal processes. Neurobiol Aging. 2019; 82: 88-101.
- Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, et al. NAD(+) in Aging: Molecular Mechanisms and Translational Implications. Trends Mol Med. 2017; 23: 899-916.
- Rizzi L, Roriz-Cruz M, Sirtuin. 1 and Alzheimer's disease: An up-to-date review. Neuropeptides. 2018; 71: 54-60.
- Van Giau V, Hulme SSA, An JP. Mitochondrial therapeutic interventions in Alzheimer's disease. J Neurol Sci. 2018; 395: 62-70.
- Fricker RA, Green EL, Jenkins SI, Griffin SM. The Influence of Nicotinamide on Health and Disease in the Central Nervous System. Int J Tryptophan Res. 2018; 11: 1178646918776658.
- Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu aRev Biochem. 2009; 78: 477-513.
- Zhou J, Xu, L, Chen, Ji F, Fortmann K, Zhang K, et al. The REGgamma-proteasome forms a regulatory circuit with IkappaBvarepsilon and NFkappaB in experimental colitis. Nat Commun. 2016; 7: 10761.
- Newton T.M, Duce JA, Bayle ED. The proteostasis network provides targets for neurodegeneration. Br J Pharmacol. 2019; 176: 3508-3514.
- Cheng J, North BJ, Zhang T, Dai X, Tao K, Guo J, Wei W. The emerging roles of protein homeostasis-governing pathways in Alzheimer's disease. Aging Cell. 2018; 17: e12801.
- Smith DM. Could a Common Mechanism of Protein Degradation Impairment Underlie Many Neurodegenerative Diseases?, J Exp Neurosci. 2018; 12: 1179069518794675.
- Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM. Synaptic Impairment in Alzheimer's Disease: A Dysregulated Symphony. Trends Neurosci. 2017; 40: 347-357.
- Zhu B, Dacso CC, O'Malley BW. Unveiling "Musica Universalis" of the Cell: A Brief History of Biological 12-Hour Rhythms. J Endocr Soc. 2018; 2: 727-752.
- Gerakis Y, Hetz C. A decay of the adaptive capacity of the unfolded protein response exacerbates Alzheimer's disease. Neurobiol Aging. 2018; 63: 162-164.
- Gerakis Y, Hetz C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer's disease, FEBS J. 2018; 285: 995-1011.









































































































































{kind=link}