Evolving Trends in the Synthesis of Deuterated Drugs for Leukemia Chemotherapy
- 1. Department of Chemistry & Biochemistry, Sharda School of Engineering & Sciences, Sharda University, India
- 2. PG Department of Chemistry, M.R.M. College (Lalit Narayan Mithila University), India
Citation
Srivastava N, Ibrahim AS, Garg U, Saxena N (2025) Evolving Trends in the Synthesis of Deuterated Drugs for Leukemia Chemotherapy. Ann Clin Pathol 12(1): 1177.
INTRODUCTION
Following the sanctioning of Deutetrabenazine (Austedo), the inaugural Deuterated pharmaceutical, there has been a notable emergence of Deuterated compounds in the therapeutic management of various diseases [1-3]. The preponderance of pharmaceuticals employed in clinical treatment is administered over a span of a few days for acute conditions, whereas the regimen extends to several months for chronic ailments. Consequently, the pharmaceutical is meticulously formulated and administered such that its concentration in the bloodstream is sustained at a constant level. In order to prolong the presence of the drug within the bloodstream, Deuterated pharmaceuticals are currently under development, which appear to exhibit greater stability and extended half-lives in comparison to their non-deuterated counterparts, thereby reducing the necessity for frequent drug administration [4]. The distinctive characteristics of deuterium facilitate enhanced precision in the modulation of drug metabolism and degradation, thereby presenting novel opportunities for the formulation of safer and more efficacious antileukemic therapeutics [5]. As the field advances, the production of deuterated pharmaceuticals, including deuterated kinase inhibitors and various targeted treatment modalities, is anticipated to yield more substantial and enduring therapeutic responses in patients afflicted with leukemia,ultimately enhancing patient outcomes and overall quality of life [6].
SIGNIFICANCE OF DEUTERIUM ATOM IN CHEMICAL STRUCTURE OF A DRUG
While the carbon-Deuterium bond is acknowledged for its enhanced strength compared to the carbon-Hydrogen bond, the interchangeability of deuterium and hydrogen atoms is frequently observed in synthetic chemical reactions [7]. The differences in bond strength can lead to what is typically referred to as the kinetic isotope effect. Kinetic isotope effects are defined as the ratio of reaction rates involving reactants that differ solely in their isotopic nature. This ratio can achieve levels of 6 to 10 fold when one or more hydrogen atoms are replaced with deuterium. Such an occurrence may affect the interactions between deuterated molecules and the enzymatic systems involved in drug metabolism. Significant effects on the pharmacokinetics (PK) of pharmaceutical agents, as opposed to their non-deuterated variants, may be identified [8]. Despite the possible disadvantages linked to deuterium substitution, including the lack of proven improvements in clinical outcomes, numerous manufacturers are currently working to bring these compounds to the market [9]. Isomers of chiral drugs might also display differences in their pharmacological characteristics, pharmacokinetics, and side effects. Since the chiral centers of these structural variants may exhibit optical instability, the incorporation of deuterium to stabilize these centers could potentially reduce or completely eliminate this instability [10]. A relevant example of this application of deuteration (deuterium-enabled chiral switching) can be seen with thalidomide, which is now approved for the acute treatment and ongoing management of the skin manifestations associated with moderate to severe erythema nodosum leprosum. The addition of deuterium acts as an extra modification to existing chemical entities in the quest for improved therapeutic efficacy while concurrently reducing adverse effects. In this case, the deuteration at the chiral center of this analog provides a significant stabilizing effect on the isomers of this substance. One isomer demonstrates marked anti-inflammatory effects in vitro, while the other isomer shows relative inactivity [11]. The Cytochrome P450 Enzyme Systems (CYPs) provide enzymes characterized by varied substrate specificities. The necessary diversity in drug metabolism is accomplished through families and subfamilies of enzymes, which generally possess wide substrate specificities and a reactive oxygenating species. When interacting with deuterated compounds, CYP reaction kinetics and pathways may be modified, potentially resulting in new metabolic products through a process known as metabolic switching [12]. A novel approach aimed at simultaneously improving the bioavailability of dextromethorphan while reducing the toxicity of quinidine involves the selective deuteration of dextromethorphan. The investigational compound AVP 786 consists of a deuterated version of dextromethorphan paired with an extremely low dose of quinidine. In a recent study, a derivative of deuterated dextromethorphan (C 10068) demonstrated antiseizure and anti-inflammatory effects in vivo, using a rat model of penetrating brain injury [13]. It is important to note that deuterium substitution does not always result in a decrease in metabolic rates. A deuterated form of paroxetine (CTP-347) has shown an increased metabolic rate compared to its non-deuterated equivalent [8]. Given that paroxetine is acknowledged for its inhibitory influence on CYP2D6, it seems that the noted enhancement in metabolism can be attributed to a reduction in this inhibitory mechanism. Although it remains ambiguous whether this phenomenon will yield a therapeutic advantage, the same investigation suggested a decrease in the interaction between paroxetine and both tamoxifen and dextromethorphan, two medications metabolized by the CYP2D6 pathway, presumably through the same mechanism [10].
Prolongation of Drug Efficacy
The substitution of hydrogen with deuterium in pharmaceutical compounds has been investigated to decelerate the metabolic conversion into less pharmacologically active metabolites or to enhance the stability of stereogenic centers, thereby prolonging the presence of active drug entities within the circulatory system [14]. This prolongation seeks to improve therapeutic efficacy and mitigate adverse effects. Although these strategies involving deuteration had remained largely theoretical for an extended period, they have recently received empirical validation with the regulatory approval of deutetrabenazine, designed for the management of motor disturbances linked to Huntington’s disease and tardive dyskinesia [15].
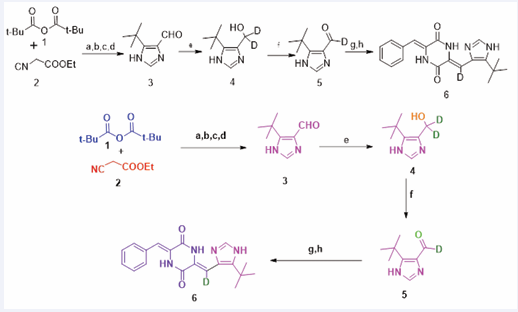
Figure 1 Synthesis of Deuterium-Substituted Plinabulin Derivative (MBRI-001). Reagents, Conditions, and Yield: (a) DBU, THF, 48 hours, 99%; (b) Formamide, 175°C, 36 hours, 45%; (c) LiAlH4, THF, 0°C to room temperature, 48 hours, 84%; (g) 1,4-diacetylpiperazine-2,5-dione, Cs2CO3, DMF, room temperature, 20 hours, 54%; (h) benzaldehyde, Cs2CO3, DMF, 50°C, 24 hours, 89%.
Modification of Physicochemical Properties and Pharmacokinetic Profiles
Deuterium, acknowledged as a stable hydrogen isotope, holds considerable relevance in medicinal chemistry due to its capacity to alter the molecular frameworks of pharmaceuticals [16]. The incorporation of deuterium into drug molecules can induce subtle modifications in their physicochemical properties, including aspects such as bond strength and bond length. Such alterations can significantly affect the pharmacological dynamics of drugs, influencing parameters like metabolic stability, binding affinity to receptors, and pharmacokinetic profiles [17]. For example, replacing hydrogen with deuterium may slow down metabolic degradation, potentially extending the drug’s half-life and augmenting its therapeutic efficacy. Furthermore, deuterated pharmaceuticals might demonstrate unique interaction patterns with biological macromolecules, enhancing selectivity and minimizing off target effects relative to their non-deuterated equivalents [18]. The principal outcome of hydrogen replacement with deuterium is the enhancement of pharmacokinetic performance, especially with respect to metrics such as half-life (t½), area under the curve (AUC), and maximum concentration (Cmax). This improvement could necessitate lower dosages or less frequent dosing to achieve adequate therapeutic exposure in patients. Nonetheless, caution is warranted, as deuteration at a particular site may redirect the compound toward alternative biotransformation pathways, potentially increasing metabolism in other regions, a phenomenon known as metabolic switching or metabolic shunting. The term “metabolic switching” aptly describes the activation of an unintended metabolic route (for example, the oxidation of an alternate site) [19].
The incorporation of deuterium atoms into pharmaceutical compounds significantly affects their pharmacokinetic properties. Unlike hydrogen, deuterium has double the atomic mass, influencing both drug metabolism and therapeutic efficacy [20]. The deuterium carbon bond is characterized by a shorter length and exhibits 6−10 times greater stability compared to its hydrogen-carbon equivalent, rendering it more resistant to cleavage. Consequently, the decreased rate of bond breakage is attributed to the Kinetic Isotope Effect (KIE). The KIE pertains to the alteration in the rate of a chemical reaction that occurs when one of the atoms in a molecular structure is substituted with its isotopic variant [21]. Due to the increased atomic weight of deuterium, C-D bonds are linked with a lower vibrational frequency and diminished zero-point energy, necessitating greater activation energy, which subsequently reduces the rate (k) of C-D bond cleavage. This rate-reducing phenomenon is identified as the Deuterium Isotope Effect (DIE) and is quantitatively expressed as kH/kD, the ratio of cleavage rates between C-H and C-D bonds. The DIE significantly influences the pharmacokinetic characteristics of various drugs metabolized via pathways dependent on C-H bond cleavage. However, slight modifications in physical attributes, such as reduced hydrophobicity and altered pKa values for acids and bases, are unlikely to markedly impact the potency or selectivity toward their biological targets [4]. The deuteration of pharmaceutical compounds reduces the metabolic rate (especially oxidative metabolism) in the gastrointestinal system or hepatic tissues, thereby promoting the absorption of the parent drug into systemic circulation or improving the bioavailability of the original agent. For instance, Deutetrabenazine, a deuterated form of tetrabenazine employed in the management of Huntington’s disease and tardive dyskinesia, exhibits enhanced metabolic stability and reduced variability in exposure, leading to a more consistent therapeutic outcome [22]. In most cases, the rate of systemic clearance remains largely unchanged. Deuterated pharmaceuticals that display this property may necessitate a lower dosage regimen and yield diminished amounts of metabolites. Considering that gastrointestinal discomfort is more significantly linked to the overall quantity of medication administered rather than its concentration within the bloodstream, this phenomenon could potentially improve patient tolerability [23].
Stabilization of Chiral Centers
Deuterium, a stable hydrogen isotope distinguished by the presence of an additional neutron, significantly affects the chemical architecture and kinetics of pharmaceutical entities [24]. The introduction of deuterium into drug formulations can bolster the stability of chiral centers by reducing the rate of enantiomerization, which denotes the transformation between enantiomers—stereoisomers that are mirror images of each other. This aspect is particularly crucial for therapeutic agents whose efficacy is contingent upon a specific enantiomer (eutomer), while the alternative enantiomer (distomer) may be ineffective or harmful. For instance, deuterium-substituted variants of thalidomide and its analogs, such as lenalidomide and avadomide, demonstrate enhanced stability and diminished rates of enantiomerization, thereby improving their pharmacological effectiveness. The Deuterium Kinetic Isotope Effect (DKIE) is instrumental in this regard, as the bond dissociation energy of the C−D bond exceeds that of the C−H bond, resulting in a decrease in bond cleavage rates and an augmentation in the stability of the pharmaceutical compounds [15].
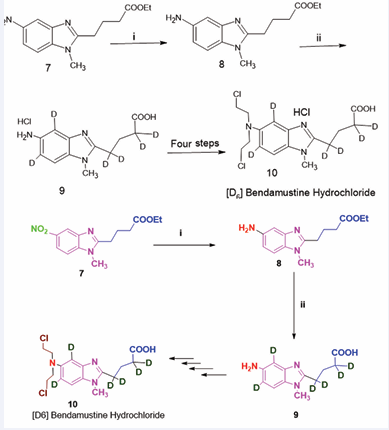
Figure 2 Synthesis of deuterium-labeled bendamustine hydrochloride
Research Applications
Deuterium-labeled pharmaceuticals play a pivotal role in scientific inquiry aimed at elucidating drug efficacy and metabolite profiles [25]. The integration of deuterium within pharmaceutical compounds facilitates meticulous tracking and analytical assessment owing to its distinctive characteristics, such as minimal natural prevalence and a unique isotopic signature. For instance, deuterium labeled therapeutics enable researchers to distinguish between various metabolic pathways and enhance their comprehension of drug interactions within biological systems [26].
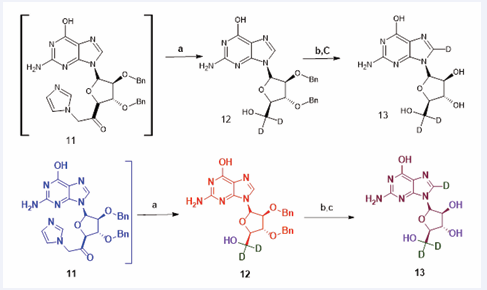
Figure 3 Synthesis of Deuterium-labeled Nelarabine Active Ingredient ([D3] ara-G) compound (13). Reagents and reaction conditions: (a) NaBD4, D2O, 18h, 50% for two steps; (b) HCOONH4, Pd/C, EtOD, reflux, 8h; (h) D2O, MW, 100°C, 1h (2 cycle), 45% for two steps.
EFFECT IN THE METABOLIC STABILITY, TOXICITY AND EXPOSURE OF DRUGS
Deuterium substitution significantly enhances metabolic stability by impeding oxidative metabolism of functional moieties such as methoxy groups, predominantly exhibiting the primary kinetic isotope effect (KIE). This modification may yield improved tolerability and dosing regimens, thereby affecting drug pharmacokinetics and the toxicological profile [15]. In essence, the modification of pharmaceuticals through deuterium exhibits the potential to influence the biological outcomes of certain drugs that undergo metabolic processes involving the cleavage of hydrogen-carbon bonds. For example, the oxidative metabolism of compounds facilitated by the cytochrome P450 enzyme, which conventionally involves the disruption of hydrogen-carbon bonds, may be affected by the presence of deuterium. Nevertheless, the kinetic isotope effect of deuterium (DIE) on the drug’s pharmacokinetics is often masked by competing in vivo factors, such as alternative metabolic pathways and differing rate-limiting steps in enzymatic reactions and biological sequestration [27]. As a result, the effects of deuteration on CYP450 mediated metabolism are complex and can vary based on the specific drug and the exact deuteration pattern; beyond its impact on metabolic rates, the introduction of deuterium may also lead to changes in the ratio of the parent drug to its metabolites, as well as fluctuations in the amounts of metabolites generated. Numerous studies have demonstrated that the incorporation of deuterium results in modified metabolite levels. Notably, the metabolites generated are congruent with those produced from their non-deuterated analogues, with the exception of the presence of deuterium, and there is no evidence to suggest that deuteration produces unique metabolites that have not been documented for the all-hydrogen counterpart. Nevertheless, diminished metabolic rates and metabolic switching, characterized by alterations in metabolite ratios, have been reported [20].
Deuteration can substantially affect the toxicity of pharmaceuticals by modifying their metabolic pathways. The introduction of deuterium into drug architectures can facilitate a reduction in the generation of toxic metabolites while simultaneously promoting the synthesis of favorable metabolites. For instance, research has indicated that deuterated compounds may display a deuterium isotope effect, potentially diminishing the incidence of toxicity associated with specific drugs. However, the magnitude of these effects is contingent upon several variables, including the particular drug, the metabolic enzymes implicated, and the dosage administered. In certain instances, even with deuterated compounds, elevated doses may still result in the production of reactive metabolites that could induce toxicity akin to that of their non-deuterated counterparts. For example, the deuterated variant of N-Nitrosodimethylamine (NDMA) demonstrated a decreased incidence of tumors in treated rats in comparison to its non-deuterated counterpart, suggesting a potential reduction in carcinogenic risk [27]. Deuteration can modify drug toxicity profiles through the modulation of metabolic pathways and tissue distribution, potentially leading to enhanced safety and tolerability when juxtaposed with non-deuterated pharmaceuticals [28].
Deuterium incorporation augments drug exposure by elevating maximum concentration (Cmax) and mean residence time (MRT) within the organism. This enhancement results in prolonged half-life and an increased area under the plasma concentration-time curve (AUC), thereby augmenting therapeutic efficacy and minimizing dosing frequency [6]. Deuterium substitution advances drug safety and efficacy by mitigating toxicity and amplifying exposure levels within target tissues. This phenomenon can culminate in a broader therapeutic window, heightened therapeutic effects, and diminished adverse effects [29].
APPLICATIONS IN MEDICINAL CHEMISTRY
Deuterium chemistry fundamentally transforms the field of medicinal chemistry by providing innovative solutions for the optimization, discovery, and development of pharmaceutical agents. The incorporation of deuterium facilitates enhancements in drug half-life and supports mechanistic investigations, thereby contributing to the advancement of therapeutics that are both safer and more effective [30]. The application of deuteration in medicinal chemistry has garnered substantial attention owing to its multifaceted benefits that augment drug characteristics and address particular challenges. It has the capacity to enhance pharmacokinetic profiles by decelerating metabolic processes, resulting in extended duration of action and diminished toxicity, particularly for compounds subject to rapid metabolic degradation [31]. Moreover, deuteration can confer stability to chiral centers in pharmaceutical compounds, leading to the preferential synthesis of targeted enantiomers, as exemplified by deuterated thalidomide derivatives that demonstrate enhanced anti-inflammatory and antitumor activities [23]. It further facilitates mechanistic examinations by enabling researchers to monitor the influence of metabolites, as illustrated in studies concerning ketamine. In addition, deuteration permits precise alterations in drug design, particularly in domains characterized by stringent structure-activity relationships, thereby optimizing drug candidates while preserving their fundamental properties. The expenses associated with deuterated reagents are becoming increasingly manageable, rendering industrial scale applications viable, and it is anticipated that the costs of enriched deuterium sources will decrease over time. The regulatory landscape has also adapted, with the FDA implementing abbreviated pathways for the approval of deuterated pharmaceuticals, which has the potential to mitigate risks and reduce expenses linked to preclinical and clinical development. Finally, deuterated compounds are utilized as internal standards in bioanalytical methodologies, thereby enhancing the precision of drug concentration measurements in biological samples [32].
RECENT DEUTERATION METHODS
The endeavor of deuterating organic compounds has attracted increased scholarly focus in recent times, primarily attributable to its potential utility in the areas of innovative drug development and synthetic chemistry. In alignment with this aim, a multitude of proficient deuterium labeling methodologies have been formulated, including hydrogen isotope exchange (HIE), reductive deuteration, and dehalogenative deuteration, all of which enable the generation of selectively deuterated compounds. In recent years, notable progress in selective isotope labeling has been achieved, culminating in a rising interest in novel strategies for the deuteration of organic molecules [33]. Over the past years, hydrogen isotope exchange (HIE) reactions have received significant attention due to the growing importance of isotope-containing molecules across diverse fields, such as materials science and the life sciences, in addition to their established role in mechanistic studies within the disciplines of chemistry and biology [34].
Hydrogen Isotope Exchange (HIE) Reactions
Hydrogen Isotope Exchange (HIE) reactions enable the integration of hydrogen isotopes at a subsequent phase within a unified synthetic framework, thus presenting a favorable substitute for conventional multistep synthesis techniques that are frequently marked by their substantial temporal and resource-intensive requirements. Moreover, HIE reactions can be perceived as pivotal processes for C–H functionalization and consequently bear considerable importance for the chemist community. Depending on the targeted outcomes, HIE reactions may necessitate either a superior level of regioselectivity or facilitate the optimal incorporation of hydrogen isotopes, and in some cases, both characteristics are sought after. In this context, metal-catalyzed HIE reactions are generally conducted using either homogeneous or heterogeneous catalytic frameworks, each of which may encounter notable constraints, such as insufficient isotope incorporation and limitations in chemo- and/or regioselectivity, respectively. Chirik’s iron-catalyzed hydrogen isotope exchange methodology utilized iron-based catalysts for hydrogen isotope exchange processes, yielding control over regioselectivity and accommodating a diverse array of substrates [28].
Photochemical Deuteration Reactions
These methodologies employ visible or ultraviolet light irradiation to activate precursor molecules, thereby facilitating the targeted incorporation of deuterium atoms into designated positions within the drug scaffold. These chemical reactions present numerous advantages, including mild reaction conditions, enhanced selectivity, and scalability. They have been effectively implemented across various categories of organic compounds, encompassing aromatic rings, alkenes, and silanes. Moreover, the advancement of metal-free and catalyst free deuteration methodologies has streamlined reaction protocols and decreased dependence on costly reagents, thereby enhancing the accessibility of deuteration [18]. Photocatalytic deuteration, often distinguished by its favorable characteristics including mild reaction conditions, superior site selectivity, and the accessibility of cost-effective deuterium sources, has surfaced as a reliable methodology for the production of organic molecules that are both medicinally and synthetically important, especially deuterated pharmaceutical agents [36].
Direct Deuteration from Heavy Water (D2 O)
This methodology involves the direct incorporation of deuterium from D2 O into organic substances. It constitutes a simplistic and cost-effective approach for deuteration, particularly beneficial for the late-stage alteration of pharmaceutical candidates. Deuterium labeling reactions are typically performed using deuterium oxide (D2 O) as the deuterium source, alongside protonated organic substrates and carbon-supported platinum-group metal catalysts. As the deuteration process advances to a sufficient degree, the deuterium concentration in D2 O decreases in tandem with the rising deuteration of organic compounds. Recently, a deuterium labeling reaction utilizing heavy water as a deuterium source within a continuous flow reactor has also been reported [37].
Enzymatic Deuteration
Enzymatic deuteration constitutes a biocatalytic methodology that entails the incorporation of deuterium (²H) into organic compounds through the utilization of enzymes, with particular emphasis on NADH-dependent reductases. This technique presents significant advantages attributable to its elevated selectivity, moderate reaction conditions, and the capacity to execute asymmetric synthesis, which is vital for the generation of chiral entities within the pharmaceutical domain [38]. Through the utilization of the functional attributes of enzymes to incorporate deuterium atoms at specific loci within pharmaceutical compounds, this approach provides exceptional regioselectivity in conjunction with gentle reaction conditions, thus making it particularly advantageous for the production of highly deuterated substances [39]. In the context of preparative scale deuteration of the chiral pharmaceutical fragment (R)-3-quinuclidinol, the biocatalytic system exhibited a noteworthy compatibility with conventional benchtop synthetic methodologies and hydrogenation techniques, facilitating the effective production of deuterated derivatives, such as a deuterated form of the incontinence medication solifenacin. This strategy not only streamlines the synthesis procedure but also permits the incorporation of deuterium concomitantly with the establishment of the chiral center, thereby obviating the necessity for subsequent hydrogen isotope exchange (HIE) [38].
SYNTHESIS OF DEUTERATED DRUGS FOR TREATMENT OF LEUKEMIA
Deutetrabenazine constitutes the preeminent deuterated therapeutic agent approved by the FDA for the management of chorea and tardive dyskinesia concomitant with Huntington’s disease. The demonstrated efficacy of deutetrabenazine has incentivized pharmaceutical companies to allocate resources towards the investigation of deuterated compounds [4]. For leukemia chemotherapy, several drugs have been deuterated, enhancing their efficacy and reducing toxicity. Some of these deuterated drugs include: Deuterated TKIs, which demonstrate improved metabolic stability and extended circulation duration within the organism, resulting in superior antileukemic effects when compared to their non deuterated equivalents [39]. The deuterated variant of dasatinib is meticulously engineered to exhibit enhanced metabolic stability and potentially improved therapeutic efficacy, thereby addressing the constraints associated with the parent compound. By mitigating the formation of deleterious metabolites and prolonging the drug’s half-life, this deuterated analog aspires to enhance the safety profile and therapeutic effectiveness of dasatinib in clinical contexts [4]. The process of deuteration modifies the pharmacokinetic characteristics of cytarabine, yielding diminished clearance rates and an extended half-life in comparison to its non-deuterated counterpart. Such alterations facilitate less frequent dosing regimens while preserving therapeutic effectiveness against leukemia cells [40]. BMS-986165, a deuterated pharmaceutical agent intended for the treatment of leukemia, commences with a methodical design and strategic planning to discern critical functional groups and positions for the incorporation of deuterium, with the objective of enhancing pharmacokinetic attributes and therapeutic effectiveness [30]. Deuterated ibrutinib potentially confers advantages such as enhanced metabolic stability and augmented therapeutic outcomes in leukemia management [22]. Below is the detail synthesis of some deuterated drugs for the Leukemia chemotherapy.
Synthesis of Deuterium-substituted Plinabulin Derivative (MBRI-001)
The derivation of the deuterium-substituted plinabulin was executed through a sequence of eight methodical synthetic procedures. This optimized synthetic route enabled the production of more than 30 grams of the target compound. The initial phase involved the formation of oxazole through the condensation reaction of ethyl isocyanoacetate and pivalic anhydride, facilitated by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) under ambient conditions for 48 hours. Subsequent purification via silica gel column chromatography allowed for the conversion of the oxazole ester into the imidazole ester through a solvolysis reaction conducted in formamide at 175°C. Following this, lithium aluminum hydride (LiAlH4 ) was utilized to reduce the imidazole ester to the corresponding alcohol, which was subsequently oxidized using manganese dioxide (MnO2 ) to produce the aldehyde. The imidazole aldehyde was then converted into the deuterium-imidazole aldehyde through a reduction reaction employing sodium borodeuteride (NaBD4 ) at -20°C, followed by an oxidation process using MnO2 The cumulative yield for the production of deuterium-imidazole aldehyde was calculated to be 24% across six synthetic stages.
A sequential aldol condensation involving two distinct aldehydes was performed on the diacetyl 2,5-piperazinedione framework, facilitated by cesium carbonate (Cs2 CO3 ) in N,N-dimethylformamide (DMF). Specifically, diacetyl-2,5-piperazinedione was reacted with the deuterium-imidazole aldehyde over a period of 20 hours at room temperature. The relatively low reactivity of the deuterium-imidazole aldehyde can be attributed to significant steric hindrance imposed by the presence of a bulky tert-butyl group at the 5-position. The subsequent condensation reaction was completed between the penultimate compound and benzaldehyde at 50°C in DMF under an inert nitrogen atmosphere. A degassed environment was employed to mitigate potential oxidation of the activated methylene within the diketopiperazine (DKP) core in the presence of Cs2 CO3 . During the final condensation reaction, a dark environment was maintained to prevent the formation of the undesired E configuration of deuterium plinabulin. The overall yield for the synthesis of this compound was quantified as 11% across the eight synthetic procedures [6].
Synthesis of deuterium-labeled bendamustine hydrochloride
A mixture comprising 4-(1-methyl-5-nitro-1H benzoimidazol-2-yl)-butyric acid ethyl ester (30.01 g, 103.02 mmol) and 10% palladium on charcoal (3.0 g), dissolved in methanol (400 mL) and ethyl acetate (200 mL), was subjected to agitation at room temperature in a hydrogen-rich environment over a period of 16 hours. The resultant solution was then filtered and concentrated under diminished pressure, yielding a white solid
In a subsequent step, a solution containing the product from the previous stage (1.02 g, 3.90 mmol) in D2 O (15 mL) was treated with 35% DCl (1.64 g, 15.32 mmol) within a 35 mL microwave reaction vessel. This vessel was sealed and subjected to heating at 180°C within a microwave synthesis system for a duration of 30 minutes. Following this heating phase, the mixture was evaporated to dryness under reduced pressure. Subsequently, fresh D2 O (15 mL) and an additional portion of 35% DCl (1.01 g, 9.34 mmol) were added to the residue. The resulting mixture underwent another heating cycle at 180°C in the microwave synthesis apparatus for an additional 30 minutes. This heating and addition process was repeated three times. The final residue was then triturated with ethanol to yield an alternative compound.
To a solution of the compound obtained from the second step (7.51 g, 27.22 mmol) in ethanol-D (60 mL), thionyl chloride was added gradually (4.84 g, 40.67 mmol) at a temperature of 0°C. The resulting mixture was maintained at 0°C for 30 minutes before being elevated to 50°C in an oil bath for a total duration of 4 hours. The advancement of the reaction was monitored using thin layer chromatography (TLC). Upon completion of the reaction, the mixture was neutralized with a saturated sodium bicarbonate solution and subsequently extracted with ethyl acetate (100 mL). The organic layers were dried over Na2 SO4 and concentrated under vacuum, resulting in the formation of a new compound.
In the final step, a solution of the compound generated in the third step (5.92 g, 22.14 mmol) was prepared in acetic acid (26.61 g, 443.2 mmol) and D2 O (49.2 g, 2.46 mol), to which oxirane (7.72 g, 175.1 mmol) was added at a temperature of 0°C. The mixture was held at 0°C for 1 hour before being raised to 25 °C in an oil bath for an extended period of 18 hours. After the reaction was complete, the mixture was neutralized with a saturated potassium carbonate solution and extracted using dichloromethane (3 × 50 mL). The organic phases were dried over Na2 SO4 and concentrated under vacuum, yielding a crude product that presented as a yellow oil. Ethyl acetate and acetone (60 mL, v/v = 3:1) were then incorporated into the crude product; after the resulting mixture underwent sonication in an ultrasonic apparatus for 30 minutes, a white solid crystallized. The product was subsequently filtered and dried at 50°C under reduced pressure, resulting in a final product characterized as a white solid
A solution containing the substance obtained from the previous step (6.01 g, 16.91 mmol) in dichloromethane (80 mL) was subjected to the gradual addition of thionyl chloride (19.3 g, 162.14 mmol) in a controlled dropwise manner at an ambient temperature of 0°C. The resulting mixture was preserved at 0°C for a period of 1 hour, followed by a gradual warming to 25°C in an oil bath over a span of 9 hours. The evolution of the reaction was meticulously monitored employing High-Performance Liquid Chromatography (HPLC). Upon the conclusion of the reaction, the solvent was eliminated through evaporation, producing the crude product (7.90 g) in the form of a viscous black oil, which was subsequently employed in the following procedure without any form of further purification.
A solution containing the product obtained from the prior step (7.90 g) was subjected to reflux in the presence of 35% DCl (53.10 g, 496 mmol) within an oil bath for a duration of 2 hours. The reaction was continuously monitored through HPLC analysis. Upon completion of the reaction, the solvent was concentrated under vacuum conditions, resulting in the crude product manifesting as a black oil. This crude product was combined with D2 O (28 mL) and allowed to stir at room temperature, facilitating the formation of a gray solid (6.32 g), which was subsequently dissolved in 35% DCl (5 mL) alongside additional D2 O (18 mL) at an elevated temperature of 60°C. Activated carbon (1.02 g) was introduced, and stirring was continued for an interval of 30 minutes. The activated carbon was subsequently removed through filtration. The resulting filtrate was evaporated to dryness. An additional 15 mL of D2 O was added to the residue, and the mixture was subjected to crystallization at 4°C for 24 hours. The precipitate was separated by filtration and rinsed with D2 O. The filter cake was then dried at 50°C under reduced pressure, yielding the final compound [22].
Synthesis of Deuterium-labeled Nelarabine Active Ingredient ([D3]ara-G)
The synthesis of compound (12) was conducted using its non-deuterated analogue (11) (520 mg, 1.118 mmol), ammonium formate (704 mg, 11.18 mmol), 10% palladium on carbon (260 mg, 0.244 mmol), and ethanol-d1 (80 mL), which were subsequently combined in a 200 mL round-bottom flask. The resulting mixture was stirred for a period of 8 hours at a temperature of 85 °C. The completion of the reaction was confirmed through Thin Layer Chromatography (TLC) analysis employing a solvent system of CH2 Cl2 /MeOH in a 5:1 ratio. After cooling to room temperature while maintaining agitation, the solid phase present within the mixture was isolated via filtration and thoroughly washed with deuterated ethanol-d1 (5mL×3). The resulting filtrate was concentrated under reduced pressure. The obtained residue was subsequently mixed with D2 O (10 mL) and subjected to microwave irradiation at 100 °C for a duration of 1 hour. Upon cooling to ambient temperature, the reaction mixture was evaporated to dryness. The residue was once again combined with D2 O (10 mL) and heated in a microwave at 100°C for an additional hour. Following cooling to room temperature, the reaction mixture was again evaporated to dryness. The resultant residue was dissolved in a 1:1 ethanol/ water mixture (80 mL), and this solution was filtered to remove any insoluble substances. The filtrate was concentrated, and the remaining residue was dried under reduced pressure, yielding the final product (13) (144 mg, 0.50mmol, yield 45%) as a white solid [40].
CONCLUSION
The incorporation of deuterium within pharmaceuticals signifies a noteworthy progression in the field of medicinal chemistry, yielding enhanced metabolic stability, diminished toxicity, and superior pharmacokinetic profiles. This assertion is substantiated by the emergence and clinical efficacy of numerous deuterated therapeutic agents. The endorsement by regulatory bodies and the commercial acquisitions of enterprises dedicated to deuterated pharmaceuticals further emphasize their economic feasibility and therapeutic promise. As methodologies for deuteration continue to advance, the development and availability of deuterated drugs are anticipated to broaden, thereby providing novel and improved therapeutic alternatives for an array of diseases, including intricate disorders such as leukemia. For instance, MBRI-001, a deuterium-substituted analogue of plinabulin, has exhibited superior pharmacokinetic characteristics and reduced toxicity in comparison to existing therapies, thereby underscoring the potential of deuterated pharmaceuticals in the enhancement of cancer treatment. Ongoing innovation in deuteration techniques and their specific applications within disease contexts will propel further advancements in medicinal chemistry, ultimately enhancing patient health outcomes.
REFERENCES
- Di Martino RMC, Maxwell BD, Pirali T. Deuterium in drug discovery: progress, opportunities and challenges. Nat Rev Drug Discov. 2023; 22: 562-584.
- Chandra Mouli HM, Vinod A, Kumari S, Tiwari AK, Kathiravan MK, Ravichandiran V, et al. Deuterated driven new chemical entities: An optimistic way to improve therapeutic efficacy. Bioorg Chem. 2023; 135: 106490.
- Dean M, Sung VW. Review of deutetrabenazine: A novel treatment for chorea associated with Huntington’s disease. Drug Des Devel Ther. 2018; 12: 313-319.
- Belete TM. Recent Updates on the Development of Deuterium- Containing Drugs for the Treatment of Cancer. Drug Des Devel Ther. 2022; 16: 3465-3472
- Koushika M, Anjana, GV. Enhancing the Properties of Natural Products and Other Drugs: Deuterium: A Novel Approach. J Nat Remed. 2024; 24.
- Ding Z, Cheng H, Wang S, Hou Y, Zhao J, Guan H. Bioorg Med Chem Lett. 2017; 27: 1416-1419.
- Nicholas Measom, Kenneth D. Down, David J. Hirst, Craig Jamieson, Eric S. Manas, Vipulkumar K, et al. Investigation of a Bicyclo[1.1.1] pentane as a Phenyl Replacement within an LpPLA2 Inhibitor. University of Strathclyde. 2016
- Russak EM, Bednarczyk EM. Impact of Deuterium Substitution on the Pharmacokinetics of Pharmaceuticals. Ann Pharmacother. 2019; 53: 211-216.
- Mullard A. Deuterated drugs draw heavier backing. Nat Rev Drug Discov. 2016; 15: 219-221.
- DeWitt S, Czarnik AW, Jacques V. Deuterium-Enabled Chiral Switching (DECS) Yields Chirally Pure Drugs from Chemically Interconverting Racemates. ACS Med Chem Lett. 2020; 11: 1789-1792.
- Hagner PR, Man HW, Fontanillo C, Wang M, Couto S, Breider M, et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood. 2015; 126: 779-789.
- Harma R, Strelevitz TJ, Gao H, Clark AJ, Schildknegt K, Obach RS, et al. Deuterium isotope effects on drug pharmacokinetics. I. System- dependent effects of specific deuteration with aldehyde oxidase cleared drugs. Drug Metab Dispos. 2012; 40: 625-634.
- Sharma R, Strelevitz TJ, Gao H, Clark AJ, Schildknegt K, Obach RS, et al. Deuterium isotope effects on drug pharmacokinetics. I. System- dependent effects of specific deuteration with aldehyde oxidase cleared drugs. Drug Metab Dispos. 2012; 40: 625-634.
- Kokel A, Kadish D, Török B. Preparation of Deuterium Labeled Compounds by Pd/C-Al-D2O Facilitated Selective H-D Exchange Reactions. Molecules. 2022; 27: 614.
- DeWitt SH, Maryanoff BE. Deuterated Drug Molecules: Focus on FDA- Approved DeutetrabenazinePublished as part of the Biochemistry series “Biochemistry to Bedside”. Biochem. 2018; 57: 472-473.
- Panda N, Uddin SKZ, Anjana GV, Ramalingam P, Palaniappan S, Raja MKMM, Kathiravan MKJ. Nat Remedies. 2023; 23: 295-305.
- Timmins GS. Deuterated drugs: where are we now? Expert Opin Ther Pat. 2014; 24: 1067-1075.
- Ranjan P, Pillitteri S, Van Der Eycken EV, Sharma UK. Photochemical methods for deuterium labelling of organic molecules. Green Chem. 2020; 22: 7725-7736.
- Horning M, Haegele K, Sommer, K, Nowlin J, Thenot, JP. (1975) CONF- 751027-1603304.
- Harbeson SL, Tung RD. Med. Deuterium Medicinal Chemistry: A New Approach to Drug Discovery and Development Chem. 2014; 24: 2-8.
- Rao N, Kini R, Kad P. Deuterated Drugs. Pharm Chem J. 2022; 55: 1372-1377.
- iu B, Qin H, Zhang Y. An efficient and facile synthesis of deuterium- labeled anticancer agent bendamustine hydrochloride. J Labelled Comp Radiopharm. 2018; 61: 869-874.
- Pirali T, Serafini M, Cargnin S, Genazzani AA. Applications of Deuterium in Medicinal Chemistry. J Med Chem. 2019; 62: 5276- 5297.
- Aubin Y, Keire DA, Marino JP, Freedberg DL. Biophysical Characterization of Proteins in Developing Biopharmaceuticals, Elsevier, 2020; 13: 375-430.
- Ou W, Qiu C, Su C, Chin J. Catal. Photo- and electro-catalytic deuteration of feedstock chemicals and pharmaceuticals: A review. 2022; 43: 956-970.
- Chang Y, Yesilcimen A, Cao M, Zhang Y, Zhang B, Chan JZ, Wasa MJ. Catalytic Deuterium Incorporation within Metabolically Stable β-Amino C-H Bonds of Drug Molecules. Am Chem Soc. 2019; 141: 14570-14575.
- Gupta M. Deuteration as a Tool for Optimization of Metabolic Stability and Toxicity of Drugs Global J Pharm Pharmacol Sci. 2017; 1: 555566.
- Koniarczyk JL, Hesk D, Overgard A, Davies IW, McNally A. A General Strategy for Site-Selective Incorporation of Deuterium and Tritium into Pyridines, Diazines, and Pharmaceuticals. J Am Chem Soc. 2018; 140: 1990-1993.
- Beumer JH, Beijnen JH, Schellens JH. Mass balance studies, with a focus on anticancer drugs. Clin Pharmacokinet. 2006; 45: 33-58.
- Cargnin S, Serafini M, Pirali T. A primer of deuterium in drug design. Future Med Chem. 2019; 11: 2039-2042.
- Wood WW. Deuterated Drugs: Isotope Distribution and Impurity Profiles. J Med Chem. 2024; 67: 16991-16999.
- Kopf S, Bourriquen F, Li W, Neumann H, Junge K, Beller M. Recent Developments for the Deuterium and Tritium Labeling of Organic Molecules. Chem Rev. 2022; 122: 6634-6718.
- Li H, Shabbir M, Li W, Lei A, Chin J. Recent Advances in Deuteration Reactions. Chem JC. 2024; 42: 1145-1156.
- Martinez-Espinar F, Salom-Català A, Bresó-Femenia E, Claver C, Baletto F, Ricart JM, et al. Bringing Selectivity in H/D Exchange Reactions Catalyzed by Metal Nanoparticles through Modulation of the Metal and the Ligand Shell. Inorg Chem. 2023; 62: 4570-4580.
- Lepron M, Daniel-Bertrand M, Mencia G, Chaudret B, Feuillastre S, Pieters G. Nanocatalyzed Hydrogen Isotope Exchange. Acc Chem Res. 2021; 54 :1465-1480.
- Zhou R, Ma L, Yang X, Cao J. Recent advances in visible-light photocatalytic deuteration reactions. Org Chem Front. 2021; 8: 426-444.
- Akutsu-Suyama K, Sajiki H, Ueda M, Asamoto M, Tsutsumi Y. Heavy water recycling for producing deuterium compounds. RSC Adv. 2022; 12: 24821-24829.
- Rowbotham JS, Ramirez MA, Lenz O, Reeve HA, Vincent KA. Bringing biocatalytic deuteration into the toolbox of asymmetric isotopic labelling techniques. Nat Commun. 2020; 11: 1454.
- Ma M, Zhao J, Cheng H, Deng M, Ding Z, Hou Y, et al. Bioorg Med Chem. 2018; 26: 4687-4692.
- Zhao D, Ren J, Wang Q, Zhang YJ. Radioanal Nucl Chem. 2020, 325,47-55.
- Howland RH. Buspirone: Back to the Future. J Psychosoc Nurs Ment Health Serv. 2015; 53: 21-24.









































































































































{kind=link}