Adrenal Gland: An Anatomical Site of Surprises: On Occasion of a Primary Leiomyosarcoma
- 1. Department of Pathology, Metaxa Cancer Memorial Hospital, Greece
- 2. Department of Surgery, Metaxa Cancer Memorial Hospital, Greece
- 3. Radiotherapy Unit, Metaxa Cancer Memorial Hospital, Greece
ABSTRACT
Primary adrenal leimyosarcoma (PLA) is an extremely rare mesenchymal adrenal tumor originating from the smooth muscle walls of the adrenal vein and its tributaries. Only thirty cases have been reported in the literature. Herein we describe a case of a 69 year-old Caucasian female who was admitted to our medical facilities reporting regular abdominal discomfort. On CT scan a right adrenal mass measuring 5.4cm x 4.3 cm was incidentally detected. The tumor was heterogeneous with a few necrotic foci. There was no evidence of extra adrenal disease. Biochemical and hormonal laboratory tests confirmed that the mass was non-functioning and the patient underwent an open right adrenalectomy. Microscopic examination revealed a mesenchymal tumor consisting of spindle shaped, glycogen-rich cells with moderate nuclear pleomorphism. On the basis of histology and immunohistochemical results a diagnosis of leiomyosarcoma was established. Postoperative adjuvant radiotherapy was administered to the patient. After one year of scheduled follow-ups, there is no evidence of local relapse or systemic disease.
KEYWORDS
Primary leiomyosarcoma ; Adrenal gland.
CITATION
Tzaida O, Papazian M, Kokkinos Ch, Provatas I, Markouizou A, et al. (2017) Adrenal Gland: An Anatomical Site of Surprises: On Occasion of a Primary Leiomyosarcoma. Ann Clin Exp Metabol 2(1): 1014
ABBREVIATIONS
PAL: Primary Adrenal Leiomyosarcoma; CT: Computed Tomography; US: Ultrasonography; MRI: Magnetic Resonance Imaging; ACTH: Adrenocorticotropic Hormone; MIBG: Metaiodobenzylguanidine Scan; HPF: High Power Fields x 40; NSAI: Non Secretory Adrenal Incidentaloma.
INTRODUCTION
Primary Adrenal Leiomyosarcoma (PAL) is an extremely rare neoplasm with only 30 cases reported in the English literature. It accounts for 0, 1– 0, 2% of all intra-abdominal soft tissue malignancies. The neoplasm is believed to originate from the smooth muscle wall of the central adrenal vein and its tributaries. A small number of PAL cases have been described in immunodeficient patients due to HIV or Eptein-Barr virus infection but the pathogenetic role of these viruses has not yet been established. Other case-reports have suggested prior trauma/injury involving the adrenal region. In the current report, we present the case of a 69-year-old female patient with a diagnosis of PAL and review the diagnostic and therapeutic challenges in the management of these tumors.
CASE REPORT
A 69 year-old Caucasian female was admitted to our medical facilities reporting regular abdominal discomfort. After abdominal CT scan a right adrenal mass measuring 5.4cm x 4.3 cm was incidentally detected. Further US, CT, and MRI scans confirmed the presence of a well-circumscribed, heterogeneous “adrenal incidentaloma” with a few necrotic foci. There was no evidence of extra adrenal disease (Figure 1).
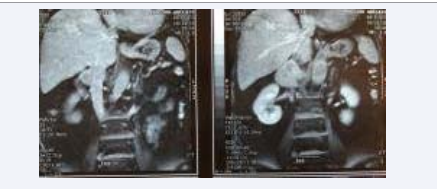
Figure 1 Right Adrenal mass on CT superomedial to the ipsilateral kidney
Biochemical and hormonal blood tests as well as serum tumor markers were within normal limits. 24-hour urine tests for cortisol and catecholamines and serum levels of aldosterone and ACTH were all normal. An MIBG scintiscan was performed with negative results, stating against the diagnosis of pheochromocytoma. Even though the tumor was non-functioning, the fact that its size exceeded 5cm leads the surgical team to the decision of performing an abdominal open right adrenalectomy. During surgical procedure the tumor was fixed against the posterior wall of the inferior vena cava (IVC) for 7cm along the course of the vessel. The adrenal vein was infiltrated (Figure 1). The tumor was successfully excised with R0 resection margins. Macroscopically the liver and abdominal viscera were unaffected. The patient recovered uneventfully and was discharged from the hospital on the 8th postoperative day.
Gross pathological examination showed a roundish, solid mass measuring 8x5, 8x 4,5cm in close vicinity to the adrenal parenchyma, surrounded by a thin fibrous pseudo capsule (Figure 2).
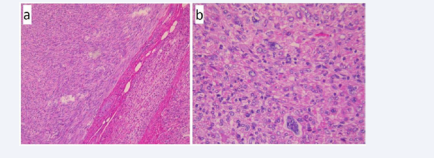
Figure 2 Microscopic examination on Hematoxylin & Eosin stain a) Neoplasm in close vicinity to the adrenal gland x100 b) Occasional bizzare cellular figures x400.
There were areas of hemorrhage and necrosis. The microscopic examination revealed a hypercellular tumor, composed of interlacing fascicles of glycogen-rich (PAS-positive), spindle-shaped cells with moderate nuclear pleiomorphism (Figure 2). Occasional bizzare features, multinucleated giant cells and focal necrosis were noted. The mitotic rate averaged between 7 to 8 mitoses per 10 HPF. In consideration of the tumor’s location, we included in the differential diagnosis tumors of the adrenal cortex and medulla, primary tumors of the liver, stromal tumors, malignant melanoma, and inflammatory myofibroblastic tumor. So we proceeded with immunohistochemical studies. On immunohistochemical analyses the neoplastic cells stained positive for vimentin, actin, desmin, HHF-35 and caldesmon (Figure 3).
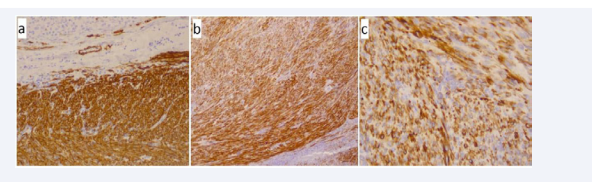
Figure 3 Immunohistochemistry a) Smooth muscle actinx100, b) Desmin x 100, c) HHF35 x 400.
CKAE1-AE3, CD34, HMB45, c-kit and S-100 protein were negative. Mitotic rate by Ki-67 was up to 30%. Based on the above findings the diagnosis of primary adrenal leiomyosarcoma was established.
Postoperatively, we decided to treat the patient with 3DCRT (3 dimensional conformal radiotherapy) to prevent loco regional relapse. A total dose of 50 Gy was administered in 25 daily fractions with a 6MV linear accelerator. After one year of regular follow-ups, our patient is healthy and shows no evidence of systemic disease or local relapse.
DISCUSSION
Retroperitoneal leiomyosarcomas are rare tumors and adrenal leiomyosarcomas even more so [1]. They are asymptomatic and therefore grow to large sizes by the time of detection; 50% are more than 15cm in diameter. Their large size, spread to surrounding structures and technical difficulties in surgical removal contribute to their poor prognosis [2]. These tumors usually arise from the walls of retroperitoneal veins most often from the inferior vena cava and show no gender predilection.
The differential diagnosis of an adrenal asymptomatic mass includes all the functional adrenal tumors, primary or metastatic adrenal malignancies, and various nonfunctioning lesions. Evaluation of a patient presenting with an adrenal incidentaloma should be directed towards determining whether the mass is hypersecretory and assessing the likelihood of malignancy (Tables 1 & 2).
PAL was firstly described by Choi and Liu in 1981 [3]. Although leiomyosarcomas are the most frequently encountered type among intra-abdominal soft tissue malignancies, only thirty cases of PAL have been reported to date [4]. Kanthan et al., reported and retrospectively analyzed 94 cases of adrenal incidentaloma, which included only one case of PAL [5]. Although the pathogenesis of this uncommon tumor has not been clearly elucidated, a few studies have suggested etiologic roles of HIV or Eptein-Barr virus [6-8].
Although the accuracy of preoperative diagnosis has improved due to advances in medical imaging, such as computed tomography (CT), magnetic resonance imaging (MRI), and ultrasonography, determination of the origin of the primary lesion can sometimes be very difficult [9,10].
In view of the rarity of this tumor and the absence of any hormonal derangement, applicable tumor serum-markers or imaging characteristics, diagnosis is based entirely on histological and immunohistochemical evaluation [11]. Convetional leiomyosarcomas show strong immmunoreactivity for smooth –muscle markers as SMA and Desmin in 90%- 95% and 70%- 90% respectively, while there is marked variability with lower percentages in the expression of these markers in pleiomorphic variant [12-14]. As our case was a typical one, all the smooth – muscle markers were positively expressed.
A variety of soft tissue tumors originated from reproperitoneum, they should be considered in the differential diagnosis of PAL (primary tumors of the adrenal cortex and medulla, metastatic tumors to adrenal gland, malignant melanoma as well as liver tumors),. For these reasons, the pathology report is critical for the classification of the tumors (classic or pleiomorphic variant) as well as for predicting the course of the disease which is usually rather aggressive with respect to its metastatic potential or frequent and early local recurrence. The survival rates seem to depend strongly on tumor size and grade, presence of necrosis, venous thrombosis, adjacent organ invasion and lymph node or distant metastases [12].
Zhou et al. reviewed the 30 cases reported in the English literature [4]. According to their data the age distribution of the patients was between 14 and 78 years, mean size of the tumor ranged from 2 to 27cm and there was no gender predilection. In twenty eight patients the tumor was unilateral and 2/30 bilateral. Four patients were immunosupressed due to HIV or Epstein-Barr infection. The most common sites for metastases were the liver, lungs and bones. Almost all the tumors were positive for smooth muscle actin (SMA), while the majority demonstrated also immunoreactivity for some of the following markers: vimentin, actin, desmin, HHF-35 and calponin. One tumor was positive for NSE and another one was reactive to CKs.
Radical surgical resection is the mainstay of therapy. Since the majority of sarcomas are surrounded by a pseudocapsule which contains neoplastic cells, this capsule is not a reliable barrier of radical resection. Therefore resection margins should be well beyond the pseudocapsule in order to prevent local recurrence. The most important prognostic factor is the ability to surgically achieve a microscopically negative margin [2]. For this reason postoperative radiotherapy is absolutely necessary for control the loco regional disease [15-18].
Currently, the only treatment of leiomyosarcoma is surgical resection because the efficacy of adjuvant therapy (chemotherapy) has not been clearly demonstrated. The disease carries a poor prognosis and a high incidence of local recurrence. The 5-year survival rate has been reported to be approximately 50% after complete en bloc resection [6,7]. Even after complete resection, the prognosis is poor and long-term observation is required.
PAL is a rare mesenchymal tumor originating from adrenals. We present such a case with typical clinico-pathological features which has been sufficiently controlled by adrenalectomy and adjuvant local radiotherapy for a 12 month-long follow up period. Although rare tumors, a high index of suspicion in non functional adrenal masses facilitates their diagnostic and therapeutic approach.
For NSAI that do not fulfil size criteria at presentation, features such as increasing size and an unfavourable imaging “phenotype” may indicate the need for surgery [6]. The lipid content of benign NSAI is high and CT “attenuation” values are significantly lower than that for adrenal carcinomas, metastases, or pheochromocytomas [7]. Noncontrasted CT attenuation values of <10 HU are typical of benign lesions, but lipid poor benign NSAI (10–40% of benign adenomas) may occasionally show higher values. Contrast washout tends to be lower (<40%) in carcinomas compared to benign adenomas. Furthermore, malignant lesions may demonstrate local invasion, necrosis, heterogeneous appearances, irregular borders, and regional lymphadenopathy [8].
ACKNOWLEDGMENTS
The authors would like to thank Mr. Konstantinos P. Karamoustos DEV MSci for his valuable support in connection with the technical processing of all the photos which are included in this project and Mrs. Antonia Papageorgiou for the English language review of the text.









































































































































{kind=link}