DNA-Hypomethylation by Hyper-Homocysteinemia: A Biochemical Mechanism Responsible for some Human Diseases, Explained to the Internist
- 1. Department of Internal Medicine and Geriatrics, “L. Vanvitelli” University of Campania, Italy
ABSTRACT
Background: DNA-hypomethylation is an epigenetic process inducing several degenerative diseases and healthy aging too.
Aim: In this review, we explained the mechanism responsible for DNA-hypomethylation and illustrated the main degenerative diseases depending from that.
Methods: The process originates by an increased homocysteine (Hcy) serum levels, that impairs S-Adenosyl-Methionine/S-Adenosyl-Homocysteine ratio (SAM/SAH ratio), also named as methylation index. This inhibits the enzymes: methyl transferases and thus, DNA methylation.
Results: The mechanisms originating from DNA-hypomethylation, as a cause of early and massive atherosclerosis, were illustrated in detail. Also mood depression, some degenerative brain’s diseases, as Alzheimer and Parkinson were significantly dependent on DNA-hypomethylation, even through these have a multifactorial origin. Some cancers and healthy aging even somehow have bound to DNA-hypomethylation.
Conclusions: DNA-hypomethylation is a fundamental biological reaction, responsible for several human diseases and due to increased Hcy levels. But, vitamins B6-9-12 supplementation, even through reduces the increased Hcy concentration, seems unable to cancel this epigenetic change.
KEYWORDS
DNA-hypomethylation ;Homocysteine ;Epigenetics ; Degenerative diseases ; Vitamins B supplementation.
CITATION
Cacciapuoti F (2018) DNA-Hypomethylation by Hyper-Homocysteinemia: A Biochemical Mechanism Responsible for some Human Diseases, Explained to the Internist. Ann Clin Exp Metabol 3(1): 1028.
ABBREVIATIONS
DNA: Desossi Nucleic Acid; CH3 : Methyl Group; Hcy: Homocysteine; CpG: Cytosine Phosphate Guanine; CpA: Cytosine Phosphate Adenine; CpT: Cytosine Phosphate Timine; CpC: Cytosine phosphate Choline; DNMTs: DNA Methyltransferases; RNA: Ribonucleic Acid; MTHFR: Methylene Tetra Hydrofolatereductase; MS: Methionine Synthase; ATP: Adenosine Tri Phosphate; SAM: S-Adenosyl Methionine; SAH: S-Adenosyl Homocysteine; MAT: Methionine Adenosyl Transferases; HHcy: Hyper-Homocysteine; VSMCs: Vascular Smooth Muscle Cells; AD: Alzheimer Disease; PD: Parkinson Disease; PRMTs: Protein Arginine Methyl Transferases; ADMA: Asymmetric di Methyl Arginine; GC: Guanine Cytosine
INTRODUCTION
INTRODUCTION DNA-methylation is a biological process essential for to initiate some degenerative diseases. It consists in the covalent transfer of a methyl group (CH3) to the cytosine ring of DNA [1]. The methyl-group can be delivered directly by dietary methyl donors including methionine, folate, betaine choline or homocysteine (Hcy). It is defined such as the process aimed to the maintenance of normal DNA structure, contributing to control gene expression and chromosomal stability. Specifically, it occurs at CpG (cytosine-phosphate-guanine) sites and results in the conversion of the cytosine to 5-methyl-cytosine. But besides the CpG site, DNA methylation can also occur at different sites, such as CpA; CpT; CpC. The addition of methyl-groups to DNA is controlled by a family of enzymes called DNA methyl-transferases (DNMTs) that are important for establishment and maintenance of the methylation process [2]. In accordance, DNA hypo-methylation caused by methylation deficiency is important too [3]. It has been proposed as a molecular marker in multiple biological processes. Particularly, DNA-hypomethylation is associated with a some key processes including genomic imprinting, atherosclerosis, neurodegenerative, autoimmune and some metabolic diseases, aging, repression of repetitive elements, and carcinogenesis. DNA-hypomethylation is able to induce changes of the genome function without changes in the DNA sequence [4]. These modifications are named “Epigenetics” and were firstly coined by Conrad Hal Waddington in 1942 [5]. The term “Epigenetics” refers to heritable changes in gene expression that does not involve changes in the underlying DNA sequence, inducing a change in phenotype without changes in genotype. Referring to the epigenetic changes, three systems there are to sustain that (DNA-methylation, histone modifications and non coding RNA).
In this review, we illustrate the mechanisms inducing DNAhypomethylation and the main pathologic processes deriving from this, bearing to mind that a key metabolite in both DNAmethylation and DNA-hypomethylation is homocysteine (Hcy) serum concentration.
HOMOCYSTEINE
Hcy is a sulphur containing an aminoacid that cannot be obtained from any dietary source, but represents an intermediate metabolite of Methionine [6,7]. Hcy in excess can be remetyilated to Methionine, by the enzyme methylene-tetra-hydrofolate reductase (MTHFR). MTHFR catalyzes the conversion of 5,10-methylene-tetra-hydrofolate to 5-methyl-tetra-hydrofolate, a co-substrate for Hcy methylation to methionine by methionine synthase (MS). On the other hand, Methionine reacts with ATP producing S-Adenosyl-Methionine (SAM), the most important methyl group donor. SAM gives a methyl groups in almost 100 known biochemical reactions and is changed to S-adenosylhomocysteine (SAH), by the enzyme methionine-adenosyltransferase (MAT). Subsequently, SAH is converted to Hcy and adenosine by SAH-hydrolase (SAH-H). This reaction is reversible therefore, the reaction proceeds in the hydrolytic direction when Hcy serum concentrations are in the normal range. On the contrary, in the presence of increased Hcy levels, it proceeds in favor of SAH. But, SAH is a potent inhibitor of SAM-dependent methylation reactions since inhibits methyl-transferases. These enzymes are responsible for methylation of some substrates, including DNA. Its inhibition induced by SAH present in excess causes DNA-hypomethylation. Concordantly, SAH excess rather than HH cyper se is responsible for DNA-hypomethylation [8].
Another route for remethylation of Hcy back to methionine exists. That uses Betaine (or Tri-Methyl-Glicine), another methyl donor that converts Hcy to Methionine. It causes both Hcy reduction and increased blood SAM levels [9,10]. Two remethylation ways, trans-methylation, trans-sulfuration pathways and methylation of substrates are schematically reported in Figure (1).
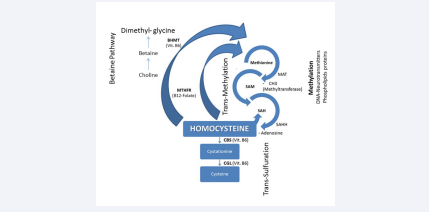
Figure 1 Two pathways for homocysteine metabolism and place for substrates’ methylation
Abbreviations: MTHFR: Methylen-Tetra-Hydro-Folate-Reductase; MAT: Methyonine-Adenosyl-Transferase; SAM: S-Adenosyl-Methyonine; SAH: S-Adenosyl-Homocysteine; SAHH: SAH-Hydrolase; CBS: Cystationine-Beta-Synthase; CGL: Cystationine-Gamma-Lyase
As previously referred, an increased Hcy level (HHcy) favors SAM synthesis. Subsequently, this compound gives a methyl group (CH3) and changes itself into SAH. The prevalence of SAH on SAM reduces trans-methylation reactions, via inhibition of methyltransferases, with consequent hypomethylation of substrates. Consequently, SAM/SAH ratio may be considered an index of trans-methylation, in other words as an indicator of methylation reactions [11-13]. In this ratio, the SAM decreased concentration is not sufficient to induce the substrates’ hypomethylation, whereas contemporary the SAH increased level is most consistently associated with reduced methylation [14]. Specifically, DNA-hypomethylation derives by the prevalence of SAH concentration on SAM levels rather than by HHcy. Therefore, SAH plasma concentration seems to be a true indicator of the inhibition DNMTs. Thus, if HHcy is directly involved as risk factor in some pathologic processes (Hcy-induced) or is a simple marker of these remain an open question [15].
In the following scheme, the SAH-inhibition of methyltransferases and DNA-hypomethylation are resumed.
inhibits ↓ methyltransferases → DNA-hypomethylation
------------------------------------------------------------------------------
In succession, the mechanisms of some human diseases deriving by DNA-hypomethylation are briefly explained [16,17].
ATHEROSCLEROSIS
Even if the mechanisms through HHcy promotes early and massive atherosclerosis are not fully understood, DNAhypomethylation is certainly included among the possible causes [8,18]. In this connection, Wang et al. have previously evidenced that HH cy inhibits the endothelial cells’ growth, demethylating cyclin A promoter via DNA-hypomethylation [19]. In other words, cyclin A demethylation will induce the inhibition of cyclin A transcription, with stop of endothelial cells’ growth, but not of vascular smooth muscle cells. Concerning this, recently Zou et al. evidenced that HHcy also act by promoting proliferation of vascular smooth muscular cells (VSMCs) in a reactive oxygen species dependent manner [20]. Jointly, these two events (stop of endothelial cells growth and proliferation of VSMC) promote degenerative atherosclerotic lesions (Figure 2).
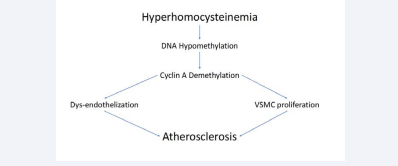
Figure 2 Atherosclerosis induced from hyperhomocysteinemia through DNA-hypomethylation (see text).
NEUROLOGIC AND PSHYCHIATRIC DISORDERS
Several evidences indicate that DNA-hypomethylation may have an important role in developing brain [21]. On the other hand, impaired SAM synthesis and, consequently, SAH prevalence on SAM may cause impaired myelin synthesis/repair with consequent demyelination, results in axonal dysfunction [22]. But, reduced Hcy re-methylation, and consequent increased SAH may lead to DNA-hypomethylation resulting in disturbed synthesis of neurotransmitters, the production of with mooddepression [23]. Concerning that, major depressive disorders were attributed to low levels of some neurotransmitters as serotonin, dopamine and norepinephrine [24].
ALZHEIMER’S DISEASE
Even if Alzheimer’s disease (AD) is a multifactorial disease, an increased Hcy concentration, with decreased SAM concentration and increased SAH levels, seems to be involved in the beginning and progression of AD [25]. In fact, the increased Hcy levels with impaired SAM/SAH ratio, can influence both brain-accumulation of β-amyloid and deposition of intracellular neurofibrillary tangles [26]. Specifically, the loss of gene methylation may alter the equilibrium among alfa, beta and gamma secretases involved of β-amyloid. In confirmation of that, experimental studies demonstrated that in AD, SAM supplementation, normalizing SAM/SAH ratio(as seen both in cell culture experiments and mouse models) can prevent these changes[27,28]. In addition, Zhuo et al., also evidenced that increased SAH concentration, in the presence both of HHcy and SAM reduction, favors brain accumulation and/or deposition of AD pathological structures [29].
PARKINSON’S DISEASE
Parkinson’s disease (PD) is a neurodegenerative disorder affecting population over the age of 65 and is characterized by loss of motor control (tremors, bradykinesia and postural instability). Non-motor symptoms include autonomic insufficiency, cognitive impairment and sleep disorders.
The brain in PD-patients is characterized by a progressive loss of neuromelanin- containing dopaminergic neurons in “substantia nigra”, with the presence of inclusions, termed as Lewy bodies (containing α-synuclein). From this point of view, several evidences indicate that DNA-hypomethylation in the substantia nigra of PD patients induces Lewy bodies’ accumulation [30]. On the other hand, the accumulation of Lewy bodies can be reduced with the administration of neurotransmitter precursor levodopa (L-Dopa) that requires SAM for its metabolism. DNA-hypomethylation is also involved in the regulation of α-synuclein gene expression, even if its specific role still remains to be further clarified. Finally, it was observed that treatment of DNA-hypomethylation with high doses of the methyl group donor SAM is effective in treating PD. In addition, L-Dopa giving causes an increase in Hcy concentration that further favors the brains’ inclusions typical of PD [31]. These are the main tangled connections between Hcy and PD.
CANCER
DNA-hypomethylation has important effects on the human genome. Among these are comprised: transcriptional repression, chromatin structure’s modulations, genomic imprinting and suppression of DNA sequences, that alters the genomic integrity [32]. These mechanisms, also called epigenetic changes, are critical components in the normal development and growth of cells and contribute to neoplastic phenotypes. In this field Robertson and Zhang found that, a lower level of leukocyte DNA- methylation is associated with many types of cancer [32,33]. Among these, colon, breast, liver, prostate and bone cancers are comprised. It was also described that global DNA-hypomethylation is implicated in the development and progression of cancer through the following mechanisms [34]:
1. DNA-hypomethylation facilitates the adaptation of cancercells to the ever- changing tumor tissue environment.
2. DNA-hypomethylation is linked to chromatin restructuring and nuclear disorganization in cancer-cells.
3. DNA-hypomethylation may occur at least partly because of cell cycle deregulation, disturbing the coordination between DNA replication and activity of DNA methyltransferases.
4. DNA-hypomethylation is in relation with the progression of tumor and metastases [35].
In addition to DNA-hypomethylation, other reactions of methylation exist and contribute to carcinogenesis. An example is the methylation of arginine 1 protein residues, catalyzed by a family of intracellular enzymes called protein arginine methyltransferases (PRMTs). They induce the asymmetric dimethylarginine (ADMA) formation. On the other hand, ADMA may control pulmonary cell behavior, even if its role in lung cancer remains elusive. Therefore, future studies are necessary to explain the mechanism through which cell types are the major contributors to altered ADMA plasma levels and by ADMA acts on lung diseases [36].
such as oxidative DNA damage, mitochondrial and nuclear genome mutations, shortening of telomeres and others [39]. Previously, several studies demonstrated that the activity of DNMT1 decreases with advance in gage [40]. So, it is believed that genome hypomethylation during ageing is a result of a passive demethylation, especially of highly methylated GC-rich DNA domain. Interestingly, accelerated DNA hypomethylation in ageing was found in individuals with PD, AD, Huntington’s disease and some viral infections. These findings suggest that DNA hypomethylation is a biomarker for aging, but also for some pathological processes ageing-related.
OTHER DISEASES
Other common diseases implicated in the epigenetic changes deriving from DNA hypomethylation are: some frequent autoimmune diseases caused by low or over activity of the immune system. Among these, rheumatoid arthritis (RA); systemic lupus erythematous (SLE) and multiple sclerosis (MS) there are. In these, DNA hypomethylation variously acts on human leukocytes antigen (HLA) and immune system. Hyperglycaemia, hyperlipidemia, obesity and shortened leukocyte telomere length are also proposed to affect DNA hypomethylation level [38].
AGING
Aging is dependent on several and interacting factors, such as oxidative DNA damage, mitochondrial and nuclear genome mutations, shortening of telomeres and others [39]. Previously, several studies demonstrated that the activity of DNMT1 decreases with advance in gage [40]. So, it is believed that genome hypomethylation during ageing is a result of a passive demethylation, especially of highly methylated GC-rich DNA domain. Interestingly, accelerated DNA hypomethylation in ageing was found in individuals with PD, AD, Huntington’s disease and some viral infections. These findings suggest that DNA hypomethylation is a biomarker for aging, but also for some pathological processes ageing-related.
CONCLUSIVE REMARKS AND FUTURE DIRECTIONS
In summary, DNA-hypomethylation dependent on Hcy cycle, with SAH prevalece on SAM, is a fundamental biochemical reaction for the normal body working. The normal SAM/SAH ratio is involved in maintaining DNA integrity, improving neurological function, preventing atherosclerotic process and is connected to nearly every biochemical processes. On the contrary, several scientific peer-reviewed articles have demonstrated that DNAhypomethylation induced by HHcy is associated with some types of cancer, metabolic ad autoimmine diseases, aging and other age-related diseases. Specifically, SAH rather than HHcy is a true risk factor for these diseases. On the contrary, HHcy may be considered as a simple biomarker of these same.
With reference to the specific treatment, it is known that deficiencies in vitamin B12, folic acid and vitamin B6 are associated with HHcy and DNA-hypomethylation (due to the SAH prevalence). In this connection, vitamins-B supplementation can modulate HHcy, SAM, SAH concentrations (and consequently SAM/SAH ratio). However, this supplementation seems to be doubtful to affect genome DNA-hypomethylation. In fact, in a double blind, randomized controlled trial, daily folic acid supplementation in subjects with moderately elevated Hcy concentration, the normalization in global DNA-methylation wasn’t found [41]. But, another study evidenced that long-term supplementation with folic acid and vitamin B12 in elderly patients with mildly elevated Hcy resulted in changes in DNA-methylation in several genes [42]. In agreement, other epidemiological studies also suggest that folate and vitamin B12 supplementation may affect DNA methylation and thereby influence genome stability, although the exact underlying mechanisms have not been clarified [43-45]. But concerning this, it must also add that both folate deficiency and excessive supplementation may equally result in abnormal DNA methylation (DNA-hypomethylation or DNA-hypermethylation) and thus affect normal gene expression [46]. Conclusively, the role of folates and other vitamins B supplementation in DNAmethylation remains uncertain for the diseases related to DNA hypomethylation, even if these nutraceutics are able to reduce elevated Hcy levels. Probably, besides the epigenetic changes, other mechanisms are involved in the pathogenesis of these degenerative diseases.
REFERENCES
3. Castro R, Rivera L, Struys EA, Jansen EE, Ravasco P, Camilo E, et al. Increased homocysteine concentration and DNA hypomethylation in vascular disease. Clin Chem. 2003; 49: 1292-1296.
10.Sunden SLF, Renduchindala MS, Park EI, Miklasz SD, Garrow T. Betaine-homocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of human gene. Arch Biochem Biophys. 1997; 345: 171-174.
32.Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001; 20: 3139-3155.
34.Craig JM, Wong NC. Epigenetics: a reference Manual Caister Academic Press. 2001.
38.Jin Z, Liu Y. DNA methylation in human diseases. Geneses Diseases. 2018; 5: 1-8.
39.Kirkwood TB. Understanding the odd science of aging. Cell. 2005; 120: 437-447.









































































































































{kind=link}