Borderline Personality: Roles of Melatonin, Oxytocin, Opioidergic System and Vagal Nerve in the Night-Time Dampening and Resetting of Mitochondria Underpin Ketamine Treatment Efficacy
- 1. CRC Scotland & London, London UK
Abstract
Borderline personality disorder (BPD) is a distressing condition with a symptomatology that includes increased sensitivity to social rejection, self-injury and subjective feeling of dysphoria. People classed with BPD often meet criteria for Major depressive disorder (MDD). BPD is still conceptualized in psychological terms as is its treatment. This article looks to define BPD on the basis of pathophysiological processes that overlap with those of depression by highlighting the role of astrocyte mitochondria dysregulation, particularly at sites of increased blood-brain barrier (BBB) permeability. The efficacy of ketamine as a fast-acting antidepressant in the treatment of MDD and BPD gives some indication as to the core changes pertinent to BPD and MDD pathophysiology. Although classically conceptualized as a post-synaptic N-methyl-d-aspartate receptor (NMDAr), antagonist, recent work shows ketamine to have direct effects on mitochondrial function. Being amphiphilic, ketamine can readily cross the BBB with astrocytes being the first CNS cell encountered. It is proposed that ketamine acts on the astrocyte mitochondrial inner membrane NMDAr. As ketamine has a short half-life, low dose ketamine has transient stress effects on mitochondrial function that leads to a preconditioning type effect by increasing mitochondrial resilience to subsequent challenges/stressors. This involves the upregulation of mitochondrial melatonin and humanin in astrocytes, which re-establish astrocyte function and also act extracellularly to dampen inflammation and oxidative stress in microglia, neurons and white matter oligodendrocytes. Pathophysiological changes in BPD (and MDD) are derived from suboptimal astrocyte mitochondrial function at sites of increased BBB permeability, including the medial prefrontal cortex (mPFC). When mPFC astrocyte metabolic function is restored, the mPFC re-establishes negative feedback on amygdala activity. Importantly, the heightened heterogeneity of BPD and MDD may be overcome by targeting this important common hub for the diverse pathophysiological processes underpinning biological heterogeneity. This has numerous future research and treatment implications.
Keywords
• Borderline Personality
• Astrocytes
• Mitochondria
• Depression
• Melatonin
• Humanin
• Ketamine
• Treatment
• Preconditioning
• Microglia
Citation
Anderson G (2026) Borderline Personality: Roles of Melatonin, Oxytocin, Opioidergic System and Vagal Nerve in the Night-Time Dampening and Resetting of Mitochondria Underpin Ketamine Treatment Efficacy. Ann Psychiatry Ment Health 14(1): 1212.
INTRODUCTION
Borderline personality disorder (BPD) is typically conceptualized as having its pathophysiological roots in early developmental processes, including prenatal, early postnatal and childhood stressors/trauma that change subsequent responses to subsequent stress [1,2]. BPD is typically defined as an increased sensitivity to perceived social rejection, whereafter dysphoria and self-injurious behaviors are more likely to develop. BPD also increases the likelihood of being classified with other poorly defined medical conditions, including Bipolar disorders [3], and addiction [4], but especially major depressive disorder (MDD). Addiction often emerges as a form of self-medication due to poorly targeted pharmaceutical treatments. BPD pathoetiology and pathophysiology have been relatively little investigated, compared to other neuropsychiatric conditions with a similar prevalence, such as schizophrenia. Consequently, BPD is psychologically defined and treated, usually with dialectical behavior therapy [5]. However, there are indicants of pathophysiological change in BPD, including suppressed serotonin and oxytocin levels [6,7]. As oxytocin is a positive allosteric modulator of the mu-opioid receptor [8], and the kappa-opioid receptor [9], a growing body of data has emerged on the role of the opioidergic system in BPD, where a decreased β-endorphin activation of the mu-opioid receptor may be coupled to an increased dynorphin activation of the kappa opioid receptor [1]. BPD pathophysiology is consistent with increased dynorphin at the kappa-opioid receptor in the basolateral amygdala, leading to feelings of dysphoria [1]. Pro-inflammatory cytokines, especially IL-6 and tumor necrosis factor (TNF)-α, are markedly increased in BPD, coupled to raised levels of oxidative stress and decreased levels of brain-derived neurotrophic factor (BDNF) [10,11] indicative of a role for inflammatory and oxidative stress processes in BPD pathophysiology. This parallels similar changes in MDD [12]. A growing body of data across diverse medical conditions has highlighted the importance of alterations in night-time dampening and resetting by melatonin and cortisol, with variations in the melatonin/cortisol ratio at night changing how body cells and systems are reset in preparation for the coming day. This is pertinent in neurodegenerative conditions such as Alzheimer’s disease [13], and amyotrophic lateral sclerosis [14], as well as cancer [15,16], cardiovascular diseases [17], and neuropsychiatric disorders [18]. Typically, an increase in pro-inflammatory cytokines increases gut permeability and circulating lipopolysaccharide (LPS), with both LPS and pro-inflammatory cytokines decreasing pineal melatonin production, thereby contributing to a heightened influence of cortisol in dampening and resetting for the coming day [13]. Gut dysbiosis and increased gut permeability are evident in BPD and therefore closely intertwined with alterations in night-time dampening and resetting [1]. The relevance of alterations in night-time dampening and resetting is most clearly highlighted over the course of aging arising from the 10-fold decrease in pineal melatonin at night between the second and ninth decade of life [19]. Suppressed pineal melatonin in the first half of sleep over aging has significant consequences for the subsequent rise in cortisol in the second half of sleep, which typically peaks around wakening, namely the cortisol awakening response (CAR), which is an integral aspect of the circadian rhythm [20]. However, many other conditions and factors can suppress pineal melatonin, including type 2 diabetes mellitus (T2DM), obesity, pro-inflammatory cytokines, stress-induced cortisol and a leaky gut/LPS as well as factors influencing the availability of the tryptophan-melatonin pathway such as hyperglycemia induced methylglyoxal [21]. T2DM, obesity, pro-inflammatory cytokines, stress induced cortisol and increased gut permeability/dysbiosis are all increased in BPD [22-24], and may function as ‘aging accelerators’ by suppressing pineal melatonin and therefore attenuating the resetting and optimization of mitochondria function during sleep [25]. Suboptimal mitochondrial function underpins the majority of raised oxidants that drive inflammatory processes, typically linked to increased NFκB, cyclooxygenase (COX)2 and induced nitric oxide synthase (iNOS) levels, as found in BPD [26], with raised NFκB showing a significant positive correlation with impulsivity scores in BPD. Overall, BPD pathophysiology may be intimately linked to processes underpinning ‘accelerated aging,’ including alterations in night-time dampening and resetting of mitochondrial function. This article therefore provides a conceptualization of BPD that involves interactions of pineal melatonin and adrenal cortisol at night in the modulation and resetting of astrocyte mitochondrial function, with significant consequences for patterned inter-area communication across the brain. This has a number of consequences for other factors and systems that can achieve resolution of inflammation, oxidative stress and psychological stress, including oxytocin, the opioidergic system and vagal nerve activation. Given the strong overlaps of BPD with MDD and the fast-acting efficacy of low-dose ketamine in the management of MDD [27], and BPD [28], the effects of ketamine on astrocyte mitochondrial function are highlighted to provide a framework to understand core processes pertinent to BPD and its strong association with stress and MDD. Next, night-time processes are briefly reviewed, including their potential relevance in BPD early developmental pathoetiology and ongoing pathophysiology.
NIGHT-TIME DAMPENING AND RESETTING
The pairing of melatonin in the first half of sleep with rising cortisol in the second half of sleep does not seem to be an evolution driven coincidence. Melatonin in the first half of sleep may dramatically influence the effects of rising cortisol in the second half of sleep. Melatonin, like gut microbiome derived butyrate, suppresses the glucocorticoid receptor (GR)-α nuclear translocation from its cytoplasmic complex with heat shock protein (hsp)90 and p23 [29,30]. Melatonin also acts on the adrenal cortex to suppress cortisol production [31]. Most data on cortisol effects have investigated the cytoplasmic GR-α. However, the GR-α may also be expressed on the plasma membrane, mitochondrial membrane and mitochondrial matrix [32], with increased cortisol acting to induce the GR-β, allowing cortisol to have diverse effects.Whether the melatonin ‘prewash’ in the first half of sleep regulates GR subtypes and sites of localization is unknown and requires experimental investigation. However, although from data restricted to the cytoplasmic GR-α, it is clear that the dramatic loss of melatonin over aging and aging-accelerating conditions can change the consequences of the subsequent cortisol rise at night that peaks in the morning CAR. Consequently, numerous diverse medical conditions are associated with accelerated aging [33-36], including BPD [37]. This is typically linked to decreased pineal melatonin and circadian dysregulation that is coupled to hypothalamic-pituitary-adrenal (HPA) axis dysregulation, as in BPD [38,39]. Aging-accelerating conditions are invariably linked to gut dysbiosis and increased gut permeability, which are mediated at least partly by the attenuation of melatonin’s maintenance of the gut barrier. This allows raised circulating LPS to suppress pineal melatonin production [40], whilst increasing adrenal cortex cortisol production, especially in the presence of obesity [41]. The suppression of pineal melatonin availability by aging accelerators therefore has a number of systemic and CNS consequences that contribute to alterations in diverse systemic processes, which ultimately act on mitochondrial function. Alterations in night-time processes pertinent to BPD are shown in Figure 1.
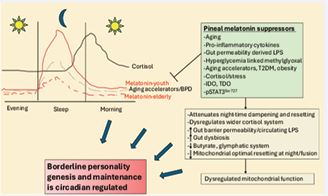
Figure 1 Night-time dampening and resetting. Shows pineal melatonin at night to be suppressed by a wide array of factors, including aging and aging accelerating conditions such as obesity, T2DM, leaky gut/LPS, and pro inflammatory cytokines. Methylglyoxal, IDO and TDO decrease tryptophan availability to initiate the tryptophan-melatonin pathway. This has consequences for CNS and systemic processes as sleep is a period where mitochondrial function is reset via melatonin driven mitochondrial fusion following daytime stressors that induce mitochondrial fission. Suboptimal mitochondrial function may be a core aspect of BPD. Abbreviations: IDO: indoleamine 2,3-dioxygenase; LPS: lipopolysaccharide; pSTAT3Ser727: phosphorylated Signal transducer and activator of transcription 3; T2DM: type 2 diabetes mellitus; TDO: tryptophan 2,3-dioxygenase.
MITOCHONDRIA AND BLOOD-BRAIN BARRIER PERMEABILITY
Recent work indicates that night-time/sleep processes prioritize resetting mitochondrial function via increased mitochondrial fusion following daytime activities and various stressors that increase mitochondrial fission [25]. This is pertinent in CNS as well as in wider systemic cells and systems. Within the CNS, suppressed pineal melatonin leads to suboptimal glymphatic system function, which is the brain’s night-time debris collection system that is driven by melatonin increasing aquaporin (AQP)4 on astrocytic end-feet [42]. Decreased pineal melatonin is also associated with increased blood-brain barrier (BBB) permeability, which is evident in BPD and in conditions of emotional dysregulation [43] as well as in the highly comorbid MDD [44,45]. As noted, BPD pathophysiology is complicated by the very common co-occurrence of MDD (up to 80%) [44], where increased BBB permeability is evident [45]. However, the overlaps of BPD and MDD likely reflect significant pathophysiological overlaps linked to alterations in stress/cortisol responsivity and attenuated pineal melatonin production at night in both conditions. Decreased glymphatic system function and increased BBB permeability are both linked to decreased pineal melatonin and may be important aspects of BPD and MDD as well as to their comorbidity.
MDD is typically associated with increased BBB permeability in the medial prefrontal cortex (mPFC), hippocampus, amygdala, thalamus and white matter, which significantly correlate with depression severity [46]. Dysregulated astrocyte functioning at these sites is accompanied, if not driven, by suboptimal mitochondrial function, as indicated by clinical and preclinical data, especially in the mPFC, showing reduced astrocyte density, altered morphology/signaling, attenuated ATP release and dysregulated Ca2+signaling [47-49]. This is most clearly shown in corticosteroid induced stress models preclinically [47], highlighting the powerful role of stress and the early developmental alterations in the wider HPA axis in both MDD [50] and BPD [51]. The mediating effect of suboptimal mitochondrial function in mPFC astrocytes in driving stress-driven mood dysregulation is supported by data showing that both optogenetic and chemogenetic activation of mPFC astrocytes increases the social rank of subordinate rodents, improves mood and mPFC excitatory/inhibitory ratio, in part by enhancing levels of excitatory amino acid transporter (EAAT) levels and therefore glutamate uptake [52]. Alterations in glutamatergic activity in the MDD PFC are therefore regulated, if not determined, by alterations to astrocyte metabolism. Investigations as to sites of increased BBB permeability in BPD still await investigation. However, imaging studies show that emotion dysregulation, deficits in social cognition, and skewed self-referential processing are associated with mPFC functional and structural alterations [53]. As the PFC negatively feeds back on heightened amygdala activity during emotion processing [54], the dysregulated function of the mPFC in BPD and MDD can lead to prolonged and heightened amygdala driven emotional influence on thought and behavioral outputs. This is typically modelled as further increasing subjective stress, leading to a maintained cortisol-linked deficit in astrocyte mitochondrial function in the mPFC [54]. The fast-acting benefits of ketamine shine some light on the core changes that may be occurring.
KETAMINE AND MITOCHONDRIA
Recent work indicates that ketamine efficacy as a fast-acting antidepressant arises from direct effects on mitochondria [55], which subsequent work proposes to be mediated by ketamine effects in astrocyte mitochondria, including via the inner mitochondrial membrane N-methyl d-aspartate receptor (NMDAr) [56]. As ketamine has a very short half-life [57] and is amphiphilic and able to cross both the plasma membrane and mitochondria bilipid membranes into mitochondria, low dose ketamine induces a mild and transient stress in mitochondria that better optimizes subsequent mitochondrial function. Ketamine may therefore precondition mitochondria to enhance their subsequent tolerance to more severe stressors/ challenges [56]. Notably, mitochondrial dysfunction is also evident in BPD and correlates with acute clinical severity [58]. Ketamine also has utility in BPD, although typically administered to alleviate the depression that highly correlates with BPD symptomatology [28]. Sites of increased BBB permeability in MDD and BPD, such as cortisol/stress driven changes in the mPFC, allow ketamine to have heightened access at these sites. As astrocytic end-feet enwrap brain endothelial cells, astrocytes are highly likely to be the first cells that ketamine encounters as it crosses into the CNS. Although astrocytes do express a plasma membrane NMDAr, this tends to be peri-synaptically localized, suggesting that ketamine entry into astrocytes is likely to result in translocation into mitochondria. By acting on the astrocyte mitochondrial inner membrane NMDAr, an initial, transient ionic dysregulation in mitochondria leads to a preconditioning ‘bounce back’ that optimize mitochondrial and astrocyte function, and therefore local neuronal regulation and the re-establishment of patterned inter-area neuronal communication. Astrocyte mitochondrial function may therefore be an important hub in both MDD and BPD, perhaps especially in the mPFC, but also in the amygdala, hippocampus, thalamus and white matter sites. These are the CNS sites upon which cortisol/stress and inflammation may act, which ketamine can reverse by preconditioning astrocyte mitochondria. Although acting directly on mitochondria, the heightened ketamine effects at sites of increased BBB permeability to optimize astrocyte function, change the inter-area patterned neuronal activity in the CNS.
REFRAMING WIDER BORDERLINE PERSONALITY DISORDER PATHOPHYSIOLOGY
Decreased pineal melatonin at night not only deprives the body and brain of a powerful antioxidant, antioxidant inducer, anti-inflammatory and optimizer of mitochondrial function [59], but also limits the capacity of melatonin to suppress the GR-α nuclear translocation as well as the melatonin induction of paraventricular nucleus (PVN) oxytocin, thereby decreasing the stimulatory effects of melatonin and oxytocin on the vagal nerve. The vagal nerve is another important mediator of dampening and resetting, including at night. Vagal stimulation by melatonin and oxytocin is in contrast to heightened cortisol levels/effects that suppress vagal nerve activation [60,61]. It is the heightened stress/cortisol activation of the GR-α in the central amygdala (CEA) that increases corticotrophin releasing hormone (CRH), which drives the induction of dynorphin and kappa-opioid receptor activation in the basolateral amygdala (BLA) leading to a state of dysphoria [62]. Melatonin (and other PVN oxytocin inducers) upregulate oxytocin induction, which activates the astrocyte oxytocin receptor in the CEA, thereby BLA preventing the cortisol/GR-α activation from upregulating the CRH/dynorphin/kappa-opioid receptor/ dysphoria pathway. Dysregulated night-time processes may be intimately linked to classical BPD pathoetiology and pathophysiology, in the amygdala, mPFC and vagal nerve. This is parsimonious with the growing body of data on the role of circadian and developmental processes in BPD [38]. Suppressed pineal melatonin also increases gut dysbiosis and gut permeability, which are evident in BPD [23]. The suppression of the gut microbiome derived short-chain fatty acid, butyrate, in the course of gut dysbiosis, also disinhibits the GR-α nuclear translocation and therefore heightens cortisol and cortisol/stress effects across CNS sites [63]. The suppression of butyrate and melatonin also has significant negative consequences for mitochondrial function [64]. Both melatonin and butyrate increase mitochondria-located sirtuin-3, which increased mitochondrial oxidative phosphorylation and decreases oxidant production by the electron transport chain [Ca65valiere et al., 2022]. Sirtuin-3 is also associated with slowing aging processes and increasing longevity, with its decrease in MDD [66], and possibly BPD, which still awaits investigation. The optimization of sirtuin-3 and mitochondrial function by melatonin and butyrate are in contrast to cortisol effects at the GR-α, as shown in other cell types [67], with sirtuin-3 therefore providing a physiological substrate on which aging-accelerating conditions may act as well as giving a physiological substrate to the putative role of cortisol/stress in BPD pathophysiology via mitochondrial alterations [58]. The suppression of melatonin and oxytocin coupled to increased cortisol/stress effects powerfully interact with the function and efficacy of the vagal nerve in BPD [68]. The vagal nerve is the parasympathetic nervous system branch of the autonomic nervous system. When stimulated, e.g., by melatonin or oxytocin, the vagal nerve releases acetylcholine (ACh), which activates a number of acetylcholine receptors, although the dampening and inflammation resolution effects of the vagal nerve may be primarily mediated via alpha-7 nicotinic acetylcholine receptor (α7nAChR) activation. α7nAChR are upregulated by pineal melatonin, indicating that pineal melatonin may not only stimulate the vagal nerve, especially via oxytocin induction, but also upregulates the immune dampening, α7nAChR [69], upon which vagal ACh acts to resolve inflammation. α7nAChR activation increases specialized proresolving mediators (SPMs) that contribute to resolution by decreasing the pro-inflammatory NF-κB dimer, p65/p50, coupled to increasing the pro-resolution NF-κB dimer, c-Rel/p50, as shown in astrocytes [70]. This shift in NF-κB dimer composition leads to the disinhibition of the mitochondrial melatonergic pathway [71], allowing released melatonin to have autocrine and paracrine effects in the local microenvironment that resolves inflammation, optimizes mitochondrial function and reestablishes homeostatic interactions. Most melatonin is produced in mitochondria, as indicated by the 98% production of melatonin in mitochondria in pinealocytes [72]. Alterations in local mitochondrial melatonin regulation may be significantly intertwined with the heightened inflammatory processes in BPD that negatively regulate mitochondrial function, as in many other medical conditions. Trans-auricular electroacupuncture stimulation of the vagal nerve to dampen gut inflammatory activity is entirely dependent upon the capacity of vagal nerve stimulation to increase local melatonin production, as shown preclinically [73], and clinically [74]. This would indicate the importance of factors acting to regulate the mitochondrial melatonergic pathway across diverse cell types in the course of distinct medical conditions, including BPD. Recent work indicates that it is the interactions of the classical intracellular signaling factors, signal transducer and activator of transcription 3 (STAT3) with NF-κB to either up- or down-regulate local melatonin production, with effects dependent upon the NF-κB dimer composition [71].
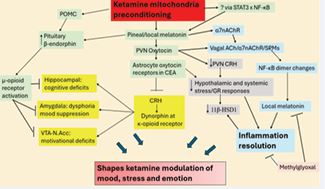
Figure 2 Ketamine’s regulation of mood, stress and emotion via mitochondrial preconditioning that upregulates the mitochondrial melatonergic pathway. Ketamine increases pineal and/or systemic melatonin, possibly via mitochondrial modulation that alters STAT3 and NF-κB interactions to upregulate melatonin as the ‘bounce back’ of preconditioning (green shade). Ketamine induction of local (PVN), and possibly pineal melatonin upregulates PVN oxytocin that activates central amygdala (CEA) astrocyte oxytocin receptors to suppress the corticotrophin-releasing hormone (CRH)/dynorphin/kappa opioid receptor pathway (yellow shade) that is induced by cortisol acting on the GR-α in the CEA. Ketamine increases POMC [76], which like melatonin, increases β-endorphin [77] that activates the mu-opioid receptor to suppress deficits in the hippocampus (cognition), amygdala (affect) and VTA/N.Acc (motivational). Acute ketamine upregulation of PVN oxytocin is therefore intimately linked to alterations in wider inter-area connectivity across the brain that are associated with dysphoria/mood, cognition and motivation regulation. Suppression of pineal or PVN melatonin and/or PVN oxytocin can disinhibit the CRH induction of the HPA axis and GR-α activation in the CEA, contributing to wider cortisol/ stress dysregulation in mood disorders as in many other medical conditions (grey shade). Increased GR-αactivation, especially in presence of the raised pro-inflammatory cytokines evident in mood disorders and BPD, enhances local cellular cortisol production by 11β-HSD1 that contributes to wider pathophysiological alterations. Consequently, the adjunctive use of melatonin and/or oxytocin can alleviate ketamine side-effects when acute ketamine is unable to upregulate pineal or PVN melatonin or oxytocin. Factors inhibiting local tryptophan-melatonin pathway induction, including methylglyoxal (orange shade), have consequences for ketamine effects in the PVN as well as for the capacity of vagal activation to achieve inflammation resolution. Abbreviations: 11β-HSD1, 11β-hydroxysteroiddehydrogenase 1? α7nAChR: alpha 7 nicotinic acetylcholine receptor; CEA: central amygdala; CRH, corticotrophin releasing hormone? GR: glucocorticoid receptor; HPA: hypothalamic-pituitary adrenal; N.Acc, nucleus accumbens? NF-κB: nuclear factor kappa-light-chain enhancer of activated B cells; POMC: pro-opiomelanocortin; PVN, hypothalamic paraventricular nucleus? SPMs: specialized proresolving mediators; STAT3: signal transducer and activator of transcription 3; VTA, ventral tegmental area.
Figure 2 shows how ketamine effects on mitochondria upregulate local melatonin, as shown in preclinical studies [75], and possibly pineal mitochondrial melatonin to regulate a plethora of physiological factors and processes linked to BPD.
Preclinical data in chronic unpredictably stressed rodents shows ketamine to increase oxytocin and oxytocin receptors [78]. Oxytocin has clinical benefits in MDD [79], and BPD [53,80], which may be at least partly mediated via oxytocin stimulating the vagal nerve [81], leading to Ch/α7nAChR/SPMs/STAT3/NF-κB/melatonin pathway in immune cells [82], including enteric glial cells [83]. Enteric glial cell α7nAChR/SPMs pathway [84], changes the composition of the NF-κB dimer [85], from a pro inflammatory NF-κB p65/p50 dimer to a NF-κB c-Rel/ p50 dimer [86], and thereby increases the likelihood of melatonin efflux in the course of local inflammation resolution in the gut. Alterations in the gut are important aspects of BPD [23], and MDD pathophysiology [87], as well as many other medical conditions, including Parkinson’s disease where increased enteric glial cell reactivity and suppressed capacity to induce the melatonergic pathway are crucial to Parkinson’s disease pathoetiology [88]. Consequently, a suppressed capacity of enteric glial cells to upregulate the melatonergic pathway stymies the beneficial effects of vagal activation by ketamine, melatonin and oxytocin in gut regulation and therefore can have significant implications for CNS processes via the gut as well as directly.
ROLE OF IL-6/JAK/STAT3 PATHWAY IN BORDERLINE PERSONALITY
As BPD is still predominantly conceptualized in psychological terms, the investigation of physiological processes has been relatively sparse, with no studies directly investigating the Janus kinase (JAK)/STAT3 pathway. However, interleukin (IL)-6 is a major inducer of the JAK/STAT3 pathway, with IL-6 being markedly increased in BPD [10], as in many other medical conditions. STAT3 is activated by phosphorylation. However, the site of STAT3 phosphorylation is of considerable importance, providing very distinct canonical (nuclear translocation), vs non-canonical (mitochondria translocating), and pSTAT3 activation consequences. Canonical pSTAT3 phosphorylation, activation and nuclear translocation follows phosphorylation of STAT3 on a specific tyrosine residue (Y705), pSTAT3Tyr705. The non-canonical pSTAT3 follows Serine727 phosphorylation and leads to the translocation of pSTAT3Ser727 to mitochondria, including within the mitochondrial matrix and to mitochondria associated membranes (MAMs) that interface the mitochondrial membrane with the endoplasmic reticulum membrane. MAMs are powerful determinants of mitochondrial Ca2+ and therefore of core changes in mitochondrial function, including apoptosis [89]. pSTAT3Ser727 is chaperoned by 14-3-3 proteins to mitochondria/MAMs, with pSTAT3Ser727 interacting with a number of mitochondrial factors, including the mitochondrial ionic regulators, Leucine zipper EF-hand containing transmembrane protein 1 (LETM1) and its parent protein LETM1 domain-containing protein 1 (LETMD1). LETM1 and LETMD1 regulate mitochondrial ionic fluxes, including Ca2+ and K+, whilst both also possess a 14-3-3 like motif [90]. In some cells, pSTAT3Ser727 forms a positive feedback loop with LETMD1 [91]. The interactions of pSTAT3Ser727 with 14-3-3 and the 14-3-3 like motif of LETM1 and LETMD1 may be important in how pSTAT3Ser727 regulates AANAT availability to initiate the mitochondrial melatonergic pathway, given that 14-3-3 is necessary for the stabilization of AANAT, in the presence of acetyl-coenzyme A (acetyl-CoA) [92]. As well as regulating the mitochondrial melatonergic pathway, pSTAT3Ser727 can also increase the mitochondrial translocation, and therefore caspase mediated activation, of the NLRP3 inflammasome to increase the pro-inflammatory cytokines, IL-1β and IL 18, as shown in other cell types [93]. The mitochondrial translocation of pSTAT3Ser727 can also increases the translocation of the pro-inflammatory NF-κB dimer component, p65, with p65 and mitochondrial NF-κB modulating mitochondrial transcription and function [93] (Figure 3). Whether the IL-6 elevation in BPD that correlates with reported childhood trauma experiences [10], also increases the pSTAT3Ser727 mitochondrial translocation will be important to determine, including in the pSTAT3 regulation of pineal AANAT/melatonin pathway, and whether this is mediated by altering 14-3 3 availability for AANAT stabilization. Local melatonergic pathway regulation is relevant across body cells and systems, such as the gut, where the vagal capacity to achieve inflammation resolution is dependent upon local melatonin upregulation and efflux [74] (Figure 3). As melatonin is derived from the tryptophan serotonin-N-acetylserotonin (NAS)-melatonin pathway, factors acting to suppress tryptophan or serotonin will also have suppressive effects on the availability of the mitochondrial melatonergic pathway. The increase in IL 6, as with other pro-inflammatory cytokines, IL-1β and tumor necrosis factor (TNF)a and especially interferon gamma (IFNg), upregulate indoleamine 2,3-dioxygenase (IDO) to convert tryptophan to kynurenine, thereby not only decreasing tryptophan availability but also activating the aryl hydrocarbon receptor (AhR), which can hydroxylate melatonin and ‘backward’ convert melatonin to its predecessor, NAS, via AhR induction of cytochrome P450 (CYP)1B1 and CYP1A2 [94]. The rise in pro inflammatory cytokines in BPD may therefore attenuate the availability of the tryptophan-melatonin pathway. Likewise, melatonin suppression, by disinhibiting the wider cortisol system, increases GR-α activation to induce tryptophan 2,3-dioxygenase (TDO) to further decrease the melatonergic pathway. The decrease in the night-time melatonin/cortisol ratio and their relative effects may therefore be contributing to alterations in the capacity of cells, microenvironments and systems to achieve resolution by attenuating the availability of the mitochondrial melatonergic pathway. Metabolic dysfunction and hyperglycemia are common in BPD, contributed to by heightened impulsivity, addiction and eating disorders [95]. Many of the detrimental effects of hyperglycemia are mediated via methylglyoxal upregulation, which is typically attributed to methylglyoxal acting as a precursor for advanced glycation end products (AGEs) that activate the receptor for AGEs (RAGE) [96]. However, methylglyoxal also has protein-protein interactions with tryptophan, thereby suppressing tryptophan availability [21]. This allows raised levels of methylglyoxal, including in astrocytes from hypertension [97], as well as in gut dysbiosis [21], to contribute to suboptimal mitochondrial function and the capacity to achieve resolution from inflammatory/ oxidative processes. This is at least partly mediated via the suppression of tryptophan for the initiation of the melatonergic pathway. Interestingly, hypertension is increased in BPD [98], suggesting a suppressed capacity to induce the mitochondrial melatonergic pathway in hypertension associated BPD, as indicated by preclinical data [21].
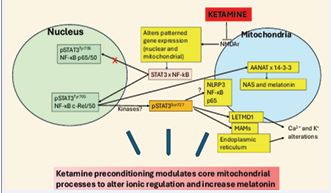
Figure 3 Ketamine preconditioning modulates core mitochondrial processes. Shows ketamine to act on the mitochondrial inner membrane NMDAr, with consequence for patterned gene expression in both mitochondria and the nucleus, including STAT3 interactions with NF-κB. In most cells, pSTAT3Tyr705 interacts with NF-κB p65/50 to induce inflammation and suppress cell melatonin production. Ketamine preconditioning is proposed to induce a pSTAT3Tyr705 interaction with NF-κB c-Rel/50, which dampens inflammatory activity and increases 14-3-3 stabilized AANAT to initiate the melatonergic pathway. Whether this is regulated by the nuclear induction of kinases to regulate pSTAT3Ser727 phosphorylation and activation requires future investigation. pSTAT3Ser727 can also translocate to MAMs to decrease endoplasmic reticulum Ca2+ influx as well as increasing LETMD1/LETM1 to regulate Ca2+ and K+. Whether such alterations in ionic regulation are aspects of ketamine’s proposed preconditioning effects awaits investigation. pSTAT3Ser727 can also increase the NLRP3 inflammasome, NF-κB and p65 translocation to mitochondria to regulate mitochondrial gene expression. Whether such pro-inflammatory/pro-apoptotic changes occur only to the toxic effects of high dose ketamine seems likely but requires future investigation. Ketamine effects on the mitochondrial melatonergic pathway may be a core aspect of its clinical utility in BPD and MDD. Abbreviations: AANAT: aralkylamine N-acetyltransferase; JAK: Janus kinase; LETM1: Leucine Zipper EF-hand containing Transmembrane protein 1; MAMs: mitochondria-associated membranes; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3: nucleotide binding domain, leucine-rich–containing family, pyrin domain–containing-3; NMDAr: N-methyl-d-aspartate receptor; STAT3: signal transducer and activator of transcription 3.
INTEGRATING BORDERLINE PERSONALITY PATHOPHYSIOLOGY
BPD pathophysiology strongly overlaps with that of MDD, including via the presence of early developmental stressors, circadian and gut microbiome alterations, suppressed tryptophan, serotonin and oxytocin, increased BBB permeability and alterations in mitochondrial function. Low dose ketamine efficacy as a fast-acting antidepressant, with utility in BPD, suggests ketamine impacts on core processes driving a depressed phenomenological state. As supported by previous literature [99], astrocyte mitochondria may be a core driver of MDD and BPD pathophysiology. This has ready logic in so much as astrocytes have a microchip-like multi-tasking capacity in contrast to the electric cable like qualities of neurons. The capacity of stress to drive alterations in astrocyte function/morphology and regulation of neurons to initiate MDD and emotional dysregulation that can be rectified by chemogenetic and photogenetic optimization of astrocyte function is strongly supportive of a crucial role for the suppression of astrocyte metabolism in depression and mood dysregulation. This may be especially relevant in the mPFC and its interface with the amygdala via the paracapsular cells of the intercalated masses [100]. Importantly, ketamine effects are not restricted to the mPFC but will be relatively enhanced at sites of BBB permeability, including the mPFC but also in the amygdala, hippocampus, thalamus, and white matter [46]. Although classically conceptualized as a post-synaptic NMDAr antagonist or negative allosteric modulator, ketamine effects arise primarily from modulating mitochondrial function [55]. As indicated above, this is likely to be primarily in astrocytes, suggesting that the suppression of astrocyte mitochondrial function may be a core aspect of MDD and BPD. The capacity of ketamine to increase local melatonin production in preclinical studies [75], suggests that ketamine will act to upregulate the astrocyte mitochondrial melatonergic pathway. It is proposed above that this may be mediated via ketamine inhibiting the NMDAr on the astrocyte inner mitochondrial membrane where it may gain entry by virtue of its amphiphilic nature and/or via organic cation transporter (OCT)3 that is expressed in astrocytes [101]. Mitochondrial NMDAr inhibition is challenging to mitochondria allowing the transient, short-life existence of ketamine to induce a ‘preconditioning’ type effect to enhance mitochondrial function, possibly involving alterations in other mitochondrial ionic regulators. Part of the preconditioning effect involves changes in the interactions of nuclear and mitochondrial pSTAT3 with NF κB composition to upregulate the astrocyte melatonergic pathway that contributes local inflammation resolution in astrocytes and cells of their local microenvironment. The putative preconditioning effects of ketamine by shifting mitochondria from a challenged to more resilient state are highly compatible with an increase in the mitochondria-derived peptide, humanin. Humanin is produced in mitochondria, predominantly within astrocytes in the CNS [102], and has intracellular and intercellular effects. Intracellular effects include the suppression of the pro-apoptotic bcl2 proteins, Bax and Bid [103,104]. Intercellular effects of humanin are mediated via at least three different plasma membrane receptors [105], including on microglia where humanin shifts microglia from an M1-like pro-inflammatory phenotype to a more quiescent M2-like phenotype, including via mitochondrial transfer from astrocytes [106]. As this shift in phenotypes in microglia [107], and macrophages [108], requires the upregulation, release and autocrine effects of melatonin, humanin effects are likely to include mitochondrial melatonergic pathway upregulation. The effects of astrocyte released humanin and melatonin also prevent demyelination and increase remyelination in white matter, damage to which has been proposed to initiate MDD [109] (Figure 4). Given the close association of BPD with MDD, the above provides a framework in which to investigate BPD pathophysiology, with ketamine effects indicating that the pharmaceutical development of a similar amphiphilic regulator of astrocyte mitochondria may over-ride the heterogenous diversity of MDD and BPD presentations by focussing on the core end-point hub on to which these heterogenous physiologies converge. The above has a number of future research and treatment implications, some of which are listed below.
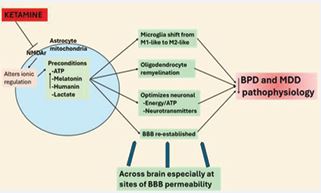
Figure 4 Ketamine’s effects highlight core changes in B D and MDD pathophysiology. Ketamine transiently inhibits the inner mitochondrial membrane NMDAr to change ionic regulation that preconditions astrocyte mitochondria to increase ATP, melatonin, humanin and lactate. Ketamine effects may be particularly evident at sites of increased BBB permeability, such as the mPFC, hippocampus and amygdala. At these sites ketamine’s induction of astrocyte melatonin and humanin efflux shifts the microglia phenotype, increases remyelination and re-establishes the BBB as well as providing neurons with lactate and re-establishing neurotransmitter regulation. This combination of effects decreases BPD and MDD symptomatology. Abbreviations: ATP: adenosine triphosphate; BBB: blood-brain barrier; BPD: borderline personality disorder; MDD: major depressive disorder; NMDAr: N-methyl-d-aspartate receptor.
FUTURE RESEARCH IMPLICATIONS
- Is the patterning of increased BBB permeability in MDD (mPFC, hippocampus, amygdala, thalamus, white matter) emulated in BPD
- Are the changes at sites of increased BBB permeability in BPD linked to stress-driven suboptimal astrocyte mitochondrial function, as shown in MDD and preclinical models [47-49]
- Does ketamine have any efficacy in BPD when criteria for comorbid MDD are not met Is efficacy mediated via preconditioning type effects in astrocyte mitochondria
- Does ketamine increase melatonin production in astrocytes, as well as other CNS and systemic cells and/or increase astrocyte humanin Antidepressant efficacy in MDD requires increased humanin release [110], indicating likely relevance in BPD.
- Is the glymphatic system suppressed in BPD If so, would the glymphatic system be increased by melatonin treatment
TREATMENT IMPLICATIONS
- Given the typically suppressed pineal melatonin release in MDD and the capacity of melatonin [111], and oxytocin [112] to enhance ketamine’s antidepressant efficacy and decrease side effects, the adjunctive use of melatonin and/or oxytocin would seem likely to better optimize treatment and increase its clinical utilization by suppressing side- effects.
- Would the adjunctive use of melatonin with ketamine attenuate ketamine’s suppression of natural killer (NK) cell levels and cytotoxicity [113] This will be important to determine given the increased risk of cancer and severe viral infection in MDD patients [114,115] and decreased longevity in BPD [116], which are linked to decreased NK cell cytotoxicity [117].
- Novel antidepressants that mimic ketamine by inhibiting the astrocyte Kir4.1 channel have recently been shown in preclinical studies to have rapid antidepressant onset effects [118].
- By reframing ketamine effects via astrocyte mitochondria, this should sharpen the focus of pharmaceutical companies to produce a ‘ketamine- like novel antidepressant,’ without side-effects, with utility in BPD.
- Ultra-low-dose buprenorphine shows efficacy in BPD symptom management [1,119], which emphasizes the relevance of opioidergic system modulation by ketamine and melatonin, as shown in Figure 2.
CONCLUSION
The mitochondrial melatonergic pathway and its regulation by night-time processes of dampening and resetting provides a physiological perspective of BPD etiology and ongoing pathophysiology. As with many other conditions linked to an early developmental etiology, such as schizophrenia and autism, the role of the mitochondrial melatonergic pathway may be intimately linked to alterations in the development of the gut and its regulation of the amygdala with consequences for alterations in stress and sensory processing driven by the early developmental effects of the amygdala on inter-area connectivity and coordination of brain regions [100, 120]. The interactions of pineal melatonin with the hypothalamus, via pineal melatonin passing through the pineal recess into the third ventricle, where it stays at a 4-fold higher concentration, vs circulatory levels, would indicate a role for night-time melatonin in the modulation of the core functions driven by hypothalamic nuclei, including via oxytocin regulation. The above provides a framework for investigating the physiological changes occurring in BPD and therefore for more targeted treatment.
REFERENCES
- Anderson G. Pathoetiology and pathophysiology of borderline personality: Role of prenatal factors, gut microbiome, mu- and kappa-opioid receptors in amygdala-PFC interactions. Prog Neuropsychopharmacol Biol Psychiatry. 2020; 98: 109782.
- Kouklidou A, Kouklidis G, Dafoulis V. A Systematic Review of Risk and Protective Factors of Borderline Personality Disorder. Cureus. 2025; 17: e85070.
- Cho M, Park C, Lee E, Park CHK. Levels and associations of borderline personality features and early maladaptive schemas in bipolar disorder: A comparative network analysis of patients with and without severe borderline personality features. J Affect Disord. 2026; 400: 121174.
- Ellappan S, Subba R, Mondal AC. Understanding borderline personality disorder: Clinical features, neurobiological insights, and therapeutic strategies. Prog Neuropsychopharmacol Biol Psychiatry. 2025; 139: 111403.
- Francis B, Fino E, Heym N. A systematic review of dialectical behaviour therapy, mentalisation-based treatment and internal family systems therapy for borderline personality disorder with comorbid depression and/or anxiety. J Psychiatr Res. 2026; 194: 221-232.
- Bocchio Chiavetto L, Tardito D, Galbiati C, Ferrari C, Lanfredi M, Pedrini L, et al. Reduction of oxytocin plasma levels in borderline personality disorder and normalization induced by psychotherapies. Psychol Med. 2025; 55: e92.
- Martial J, Paris J, Leyton M, Zweig-Frank H, Schwartz G, Teboul E, et al. Neuroendocrine study of serotonin function in female borderline personality disorder patients: a pilot study. Biol Psychiatry. 1997; 42: 737-739.
- Meguro Y, Miyano K, Hirayama S, Yoshida Y, Ishibashi N, Ogino T, et al. Neuropeptide oxytocin enhances μ opioid receptor signaling as a positive allosteric modulator. J Pharmacol Sci. 2018; 137: 67-75.
- Miyano K, Yoshida Y, Hirayama S, Takahashi H, Ono H, Meguro Y, et al. Oxytocin Is a Positive Allosteric Modulator of κ-Opioid Receptors but Not δ-Opioid Receptors in the G Protein Signaling Pathway. Cells. 2021; 10: 2651.
- Spohrs J, Kühnle V, Reber SO, Mikusky D, Sanhüter N, Macchia A, et al. The role of the endocannabinoid system in the interplay of adverse childhood experiences and interleukin 6 in individuals with borderline personality disorder. Psychopharmacology (Berl). 2025.
- Forte ARCC, Lessa PHC, Chaves Filho AJM, Aquino PEA, Brito LM, Pinheiro LC, et al. Oxidative stress and inflammatory process in borderline personality disorder (BPD): a narrative review. Braz J Med Biol Res. 2023; 56: e12484.
- Anderson G, Maes M. Oxidative/nitrosative stress and immuno- inflammatory pathways in depression: treatment implications. Curr Pharm Des. 2014; 20: 3812-3847.
- Anderson G. Physiological processes underpinning the ubiquitous benefits and interactions of melatonin, butyrate and green tea in neurodegenerative conditions. Melatonin Res. 2024; 7: 20-46.
- Anderson G. Amyotrophic Lateral Sclerosis Pathoetiology and Pathophysiology: Roles of Astrocytes, Gut Microbiome, and Muscle Interactions via the Mitochondrial Melatonergic Pathway, with Disruption by Glyphosate-Based Herbicides. Int J Mol Sci. 2022; 24: 587.
- Anderson G. Melatonin, BAG-1 and cortisol circadian interactions in tumor pathogenesis and patterned immune responses. Explor Target Antitumor Ther. 2023; 4: 962-993.
- Anderson G. Gut Microbiome and Circadian Interactions with Platelets Across Human Diseases, including Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Cancer. Curr Top Med Chem. 2023b; 23: 2699-2719.
- Anderson G. Diabetic cardiomyopathy: roles of STAT3 interactions with NF-kB dimer composition in the modulation of the cardiac mitochondrial melatonergic pathway. AIMS Mol Sci. 2025; 12: 385-410.
- Anderson G. Autism Pathoetiology and Pathophysiology: Roles of STAT3 and NF-κB Dimer Interactions in Regulating the Mitochondrial Melatonergic Pathway in Placental, CNS, and Systemic Cells. Front Biosci (Landmark Ed). 2026; 31: 46455.
- Karasek M. Melatonin, human aging, and age-related diseases. ExpGerontol. 2004; 39: 1723-1729.
- Velazquez Sanchez C, Dalley JW. The cortisol awakening response:Fact or fiction Brain Neurosci Adv. 2025; 9: 23982128251327712.
- Md Samsuzzaman, Hong SM, Lee JH, Park H, Chang KA, Kim HB, et al. Depression like-behavior and memory loss induced by methylglyoxal is associated with tryptophan depletion and oxidative stress: a new in vivo model of neurodegeneration. Biol Res. 2024; 57: 87.
- Powers AD, Oltmanns TF. Borderline personality pathology and chronic health problems in later adulthood: the mediating role of obesity. Personal Disord. 2013; 4: 152-159.
- Rössler H, Flasbeck V, Gatermann S, Brüne M. Alterations of the gut microbiota in borderline personality disorder. J Psychosom Res. 2022; 158: 110942.
- Kahl KG, Bens S, Ziegler K, Rudolf S, Dibbelt L, Kordon A, et al. Cortisol, the cortisol-dehydroepiandrosterone ratio, and pro-inflammatory cytokines in patients with current major depressive disorder comorbid with borderline personality disorder. Biol Psychiatry. 2006; 59: 667-671.
- Richardson RB, Mailloux RJ. Mitochondria Need Their Sleep: Redox, Bioenergetics, and Temperature Regulation of Circadian Rhythms and the Role of Cysteine-Mediated Redox Signaling, Uncoupling Proteins, and Substrate Cycles. Antioxidants (Basel). 2023; 12: 674.
- MacDowell KS, Marsá MD, Buenache E, Villatoro JML, Moreno B, Leza JC, et al. Inflammatory and antioxidant pathway dysfunction in borderline personality disorder. Psychiatry Res. 2020; 284: 112782.
- Guo H, Tang L, He M, Tang W, Liu J, Wu S, et al. Comparative safety and tolerability of ketamine and esketamine for major depressive disorder: a systematic review and meta-analysis. Front Pharmacol. 2025; 16: 1681060.
- Liester M, Wilkenson R, Patterson B, Liang B. Very Low-Dose Sublingual Ketamine for Borderline Personality Disorder and Treatment-Resistant Depression. Cureus. 2024; 16: e57654.
- Quiros I, Mayo JC, Garcia-Suarez O, Hevia D, Martin V, Rodríguez C, et al. Melatonin prevents glucocorticoid inhibition of cell proliferation and toxicity in hippocampal cells by reducing glucocorticoid receptor nuclear translocation. J Steroid Biochem Mol Biol. 2008; 110: 116- 124.
- Kim MJ, Choi GE, Chae CW, Lim JR, Jung YH, Yoon JH, et al. Melatonin-mediated FKBP4 downregulation protects against stress- induced neuronal mitochondria dysfunctions by blocking nuclear translocation of GR. Cell Death Dis. 2023; 14: 146.
- Torres-Farfan C, Richter HG, Rojas-García P, Vergara M, Forcelledo ML, Valladares LE, et al. mt1 Melatonin receptor in the primate adrenal gland: inhibition of adrenocorticotropin-stimulated cortisol production by melatonin. J Clin Endocrinol Metab. 2003; 88: 450-458.
- Zhao X, Huang X, Huang C, Wang X, Yang Y, Dang R, et al. Study on the mechanism of glucocorticoid receptor mitochondrial translocation and glucocorticoid-induced apoptosis in macrophages. Immunopharmacol Immunotoxicol. 2024; 46: 482-495.
- Fraszczyk E, Thio CHL, Wackers P, Dollé MET, Bloks VW, Hodemaekers H, et al. DNA methylation trajectories and accelerated epigenetic aging in incident type 2 diabetes. Geroscience. 2022; 44: 2671-2684.
- Hu L, Li J, Tang Z, Gong P, Chang Z, Yang C, et al. How does biological age acceleration mediate the associations of obesity with cardiovascular disease Evidence from international multi-cohort studies. Cardiovasc Diabetol. 2025; 24: 209.
- Qi D, Liu P, Wang Y, Tai X, Ma S, Wang Y. A multi-omic dissection of molecular hallmarks of accelerated aging in schizophrenia. Sci Rep. 2025; 15: 17856.
- Fries GR, Garza S, Zhao NO, Bass AW, Lima CNC, Kobori N, et al. Association between epigenetic aging acceleration and amyloid biomarkers in bipolar disorder. medRxiv [Preprint]. 2025: 2025.04.06.25325186.
- Boström ADE, Andersson P, Jamshidi E, Wilczek A, Nilsonne Å, Rask- Andersen M, et al. Accelerated epigenetic aging in women with emotionally unstable personality disorder and a history of suicide attempts. Transl Psychiatry. 2023; 13: 66.
- McGowan NM, Saunders KEA. The Emerging Circadian Phenotype of Borderline Personality Disorder: Mechanisms, Opportunities and Future Directions. Curr Psychiatry Rep. 2021; 23: 30.
- Kulakova E, Graumann L, Wingenfeld K. The Hypothalamus-Pituitary- Adrenal Axis and Social Cognition in Borderline Personality Disorder. Curr Neuropharmacol. 2024; 22: 378-394.
- da Silveira Cruz-Machado S, Pinato L, Tamura EK, Carvalho-Sousa CE, Markus RP. Glia-pinealocyte network: the paracrine modulation of melatonin synthesis by tumor necrosis factor (TNF). PLoS One. 2012; 7: e40142.
- Vakharia K, Hinson JP. Lipopolysaccharide directly stimulates cortisol secretion by human adrenal cells by a cyclooxygenase-dependent mechanism. Endocrinology. 2005; 146: 1398-1402.
- Sun H, Cao Q, He X, Du X, Jiang X, Wu T, et al. Melatonin Mitigates Sleep Restriction-Induced Cognitive and Glymphatic Dysfunction Via Aquaporin-4 Polarization. Mol Neurobiol. 2025; 62: 11443-11465.
- Ocab O, Saad HA, Mashal R, Osama K, Marzouk M, Hamdi N. Influence of feelings on the blood-brain barrier (BBB) and drug delivery. Prog Brain Res. 2025; 293: 203-242.
- Rao S, Broadbear J. Borderline personality disorder and depressivedisorder. Australas Psychiatry. 2019; 27: 573-577.
- Wu L, Wu F, Li M, Liu Y, Wang S, Guo S, et al. The elevated plasma levels of Claudin-5 are associated with peripheral inflammatory activity in patients with major depressive disorder. J Affect Disord. 2026; 398: 120988.
- Shang B, Wang T, Zhao S, Yi S, Zhang T, Yang Y, et al. Higher Blood- brain barrier permeability in patients with major depressive disorder identified by DCE-MRI imaging. Psychiatry Res Neuroimaging. 2024; 337: 111761.
- González-Arias C, Sánchez-Ruiz A, Esparza J, Sánchez-Puelles C, Arancibia L, Ramírez-Franco J, et al. Dysfunctional serotonergic neuron-astrocyte signaling in depressive-like states. Mol Psychiatry. 2023; 28: 3856-3873.
- Puentes-Orozco M, Albarracin SL, Velásquez MM. Neuroinflammation and major depressive disorder: astrocytes at the crossroads. Front Cell Neurosci. 2024; 18: 1504555.
- Si X, Miguel-Hidalgo JJ, O’Dwyer G, Stockmeier CA, Rajkowska G. Age- dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression. Neuropsychopharmacology. 2004; 29: 2088-2096.
- Jiang L, Xue L, Juruena MF. The impact of early life stress on the hypothalamic-pituitary-adrenal axis in unipolar major depression: A systematic review. Psychoneuroendocrinology. 2025; 181: 107607.
- Thomas N, Gurvich C, Kulkarni J. Borderline personality disorder, trauma, and the hypothalamus-pituitary-adrenal axis. Neuropsychiatr Dis Treat. 2019; 15: 2601-2612.
- Noh K, Oh J, Cho WH, Hwang M, Lee SJ. Astrocyte-derived dominance winning reverses chronic stress-induced depressive behaviors. Mol Brain. 2024; 17: 59.
- Giannoulis E, Nousis C, Sula IJ, Georgitsi ME, Malogiannis I. Understanding the Borderline Brain: A Review of Neurobiological Findings in Borderline Personality Disorder (BPD). Biomedicines. 2025; 13: 1783.
- Berboth S, Morawetz C. Amygdala-prefrontal connectivity during emotion regulation: A meta-analysis of psychophysiological interactions. Neuropsychologia. 2021; 153: 107767.
- Yue C, Wang N, Zhai H, Yuan Z, Cui Y, Quan J, et al. Adenosine signalling drives antidepressant actions of ketamine and ECT. Nature. 2026; 649: 423-431.
- Anderson G. Low dose ketamine preconditions astrocyte mitochondria to achieve antidepressant efficacy via adenosine, humanin and melatonin upregulation and efflux. 2026
- Clements JA, Nimmo WS, Grant IS. Bioavailability, pharmacokinetics, and analgesic activity of ketamine in humans. J Pharm Sci. 1982; 71: 539-542.
- Behnke A, Rappel M, Ramo-Fernández L, Mavio?lu RN, Weber B, Neuner F, et al. Mitochondrial respiratory activity and DNA damage in peripheral blood mononuclear cells in borderline personality disorder. Psychol Med. 2025; 55: e365.
- Anderson G, Maes M. Local melatonin regulates inflammation resolution: a common factor in neurodegenerative, psychiatric and systemic inflammatory disorders. CNS Neurol Disord Drug Targets. 2014; 13: 817-827.
- Llewellyn-Smith IJ, Kellett DO, Jordan D, Browning KN, Travagli RA. Oxytocin-immunoreactive innervation of identified neurons in the rat dorsal vagal complex. Neurogastroenterol Motil. 2012; 24: e136- 146.
- Pellissier S, Dantzer C, Mondillon L, Trocme C, Gauchez AS, Ducros V, et al. Relationship between vagal tone, cortisol, TNF-alpha, epinephrine and negative affects in Crohn’s disease and irritable bowel syndrome. PLoS One. 2014; 9: e105328.
- Anderson G. Polycystic Ovary Syndrome Pathophysiology: Integrating Systemic, CNS and Circadian Processes. Front Biosci (Landmark Ed). 2024; 29: 24.
- Zhao X, Zhu M, Wang Z, Gao M, Long Y, Zhou S, et al. The Alleviative Effect of Sodium Butyrate on Dexamethasone-Induced Skeletal Muscle Atrophy. Cell Biol Int. 2025; 49: 508-521.
- Anderson G, Maes M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr Top Med Chem. 2020; 20: 524-539.
- Cavaliere G, Catapano A, Trinchese G, Cimmino F, Penna E, Pizzella A, et al. Butyrate Improves Neuroinflammation and Mitochondrial Impairment in Cerebral Cortex and Synaptic Fraction in an Animal Model of Diet-Induced Obesity. Antioxidants (Basel). 2022; 12: 4.
- Chen CY, Wang YT, Liu LJ, Zhang Y. Sirtuin 3, a mitochondrial metabolic enzyme, links the mitochondrial function to neurophysiology in depression. Prog Neuropsychopharmacol Biol Psychiatry. 2025; 143: 111563.
- Chen L, Wang BZ, Xie J, Zhang RY, Jin C, Chen WK, et al. Therapeutic effect of SIRT3 on glucocorticoid-induced osteonecrosis of the femoral head via intracellular oxidative suppression. Free Radic Biol Med. 2021; 176: 228-240.
- Guerriero G, Liljedahl SI, Carlsen HK, López Muñoz M, Daros AR, Ruocco AC, et al. Transcutaneous auricular vagus nerve stimulation to acutely reduce emotional vulnerability and improve emotional regulation in borderline personality disorder (tVNS-BPD): study protocol for a randomized, single-blind, sham-controlled trial. Trials. 2024; 25: 397
- Markus RP, Silva CL, Franco DG, Barbosa EM Jr, Ferreira ZS. Is modulation of nicotinic acetylcholine receptors by melatonin relevant for therapy with cholinergic drugs Pharmacol Ther. 2010; 126: 251-262.
- Kaya Z, Belder N, Sever-Bahçekap?l? M, Erdener ?E, Dönmez-Demir B, Ba?c? C, et al. Spreading depolarization triggers pro- and anti- inflammatory signalling: a potential link to headache. Brain. 2025: awaf015.
- Córdoba-Moreno MO, Santos GC, Muxel SM, Dos Santos-Silva D, Quiles CL, Sousa KDS, et al. IL-10-induced STAT3/NF-κB crosstalk modulates pineal and extra-pineal melatonin synthesis. J Pineal Res. 2024; 76: e12923.
- Moravcová S, Filipovská E, Spišská V, Svobodová I, Novotný J, BendováZ. The Circadian Rhythms of STAT3 in the Rat Pineal Gland and Its Involvement in Arylalkylamine-N-Acetyltransferase Regulation. Life (Basel). 2021; 11: 1105.
- Zhang Y, Zou N, Xin C, Wang Y, Zhang Z, Rong P, Li S. Transcutaneous auricular vagal nerve stimulation modulates blood glucose in ZDF rats via intestinal melatonin receptors and melatonin secretion. Front Neurosci. 2024; 18: 1471387.
- Li J MD, Wang L MD, Zhang Y MD, Meng R MD, Zhu B MD, Chen JDZ PHD, et al. Transcutaneous auricular vagal nerve stimulation improves functional dyspepsia with sleep disturbance via enhanced vagal activity: a randomized controlled trial. Int J Surg. 2026; 112: 961-972.
- Pang CS, Mulnier C, Pang SF, Yang JC. Effects of halothane, pentobarbital and ketamine on serum melatonin levels in the early scotophase in New Zealand white rabbits. Biol Signals Recept. 2001; 10: 310-316.
- Jiang C, DiLeone RJ, Pittenger C, Duman RS. The endogenous opioid system in the medial prefrontal cortex mediates ketamine’s antidepressant-like actions. Transl Psychiatry. 2024; 14: 90.
- Shavali S, Ho B, Govitrapong P, Sawlom S, Ajjimaporn A, Klongpanichapak S, et al. Melatonin exerts its analgesic actions not by binding to opioid receptor subtypes but by increasing the release of beta-endorphin an endogenous opioid. Brain Res Bull. 2005; 64: 471-479.
- Edem EE, Oguntala OA, Ikuelogbon DA, Nebo KE, Fafure AA, Akinluyi ET, et al. Prolonged ketamine therapy differentially rescues psychobehavioural deficits via modulation of nitro-oxidative stress and oxytocin receptors in the gut-brain-axis of chronically-stressed mice. Psychoneuroendocrinology. 2023; 158: 106370.
- Wang M, Hou S, Tian C, Fu Z, Jie J. Effects of Intranasal Oxytocin Across Various Depressive Disorders. Psychiatry Investig. 2025; 22: 964-978.
- Giannoulis E, Nousis C, Eytaxia LA, Kaimakami O, Malogiannis I. The Promise of Intranasal Oxytocin in Treating Borderline Personality Disorder: A Narrative Review. Brain Sci. 2025; 15: 708.
- Nowacka A, ?niegocki M, Zió?kowska EA. Vagal Oxytocin Receptors as Molecular Targets in Gut-Brain Signaling: Implications for Appetite, Satiety, Obesity, and Esophageal Motility-A Narrative Review. Int J Mol Sci. 2025; 26: 7812.
- Merchant W, Wyler S, Chen B, Gautron L. The molecular components of the anti-inflammatory cholinergic pathway are extrasplenic. PLoS One. 2025; 20: e0331707.
- Costantini TW, Krzyzaniak M, Cheadle GA, Putnam JG, Hageny AM, Lopez N, et al. Targeting α-7 nicotinic acetylcholine receptor in the enteric nervous system: a cholinergic agonist prevents gut barrier failure after severe burn injury. Am J Pathol. 2012; 181: 478-486.
- Huang Y, Dong S, Li X, Shi J, Zhang Y, Liu S, et al. VNS-mediated α7nAChR signaling promotes SPM synthesis via regulation of netrin-1 expression during LPS-induced ALI. FASEB J. 2024; 38: e9664.
- Kim J, Park HM, Lim CM, Jeon KB, Kim S, Lee J, et al. Specialized pro- resolving mediator 7S MaR1 inhibits IL-6 expression via modulating ROS/p38/ERK/NF-κB pathways in PM10-exposed keratinocytes. BMB Rep. 2024; 57: 490-496.
- Barbosa MLL, Costa DVDS, de Pacífico DM, Rebouças CDSM, Warren CA, de Leitão RFC, et al. Role of TLR4 in Enteric Glia Response to Clostridioides Difficile Toxins: Insights From In Vivo and In Vitro Studies. J Cell Mol Med. 2025; 29: e70943.
- Slyepchenko A, Maes M, Machado-Vieira R, Anderson G, Solmi M, Sanz Y, et al. Intestinal Dysbiosis, Gut Hyperpermeability and Bacterial Translocation: Missing Links Between Depression, Obesity and Type 2 Diabetes. Curr Pharm Des. 2016; 22: 6087-6106.
- Anderson G. Enteric glial cell and astrocyte melatonin regulation in Parkinson disease pathogenesis and pathophysiology. Academia Biology. 2026; 4.
- Su Y, Huang X, Huang Z, Huang T, Xu Y, Yi C. STAT3 Localizes in Mitochondria-Associated ER Membranes Instead of in Mitochondria. Front Cell Dev Biol. 2020; 8: 274.
- Lupo D, Vollmer C, Deckers M, Mick DU, Tews I, Sinning I, et al. Mdm38 is a 14-3-3-like receptor and associates with the protein synthesis machinery at the inner mitochondrial membrane. Traffic. 2011; 12: 1457-1466.
- Zhang JL, Liu XZ, Wang PY, Chen GW, Jiang Y, Qiao SK, et al. Targeting HCCR expression resensitizes gastric cancer cells to chemotherapy via down-regulating the activation of STAT3. Sci Rep. 2016; 6: 24196.
- Obsil T, Ghirlando R, Klein DC, Ganguly S, Dyda F. Crystal structure of the 14-3-3 zeta:serotonin N-acetyltransferase complex. a role for scaffolding in enzyme regulation. Cell. 2001; 105: 257-267.
- Luo L, Wang F, Xu X, Ma M, Kuang G, Zhang Y, et al. Author Correction: STAT3 promotes NLRP3 inflammasome activation by mediating NLRP3 mitochondrial translocation. Exp Mol Med. 2024; 56: 2097.
- Mokkawes T, Lim ZQ, de Visser SP. Mechanism of Melatonin Metabolism by CYP1A1: What Determines the Bifurcation Pathways of Hydroxylation versus Deformylation J Phys Chem B. 2022; 126: 9591-9606.
- Bozzatello P, Marin G, Gabriele G, Brasso C, Rocca P, Bellino S. Metabolic Dysfunctions, Dysregulation of the Autonomic Nervous System, and Echocardiographic Parameters in Borderline Personality Disorder: A Narrative Review. Int J Mol Sci. 2024; 25: 12286.
- Jiang W, Gong M, Shen L, Yu C, Ruan H, Chen P, et al. The Receptor for Advanced Glycation End-products in the Mouse Anterior Cingulate Cortex is Involved in Neuron?Astrocyte Coupling in ChronicInflammatory Pain and Anxiety Comorbidity. Mol Neurobiol. 2025;62: 7183-7204.
- Zhang WY, Zhao CM, Wang CS, Xie X, Li YQ, Chen BB, et al. Methylglyoxal accumulation contributes to accelerated brain aging in spontaneously hypertensive rats. Free Radic Biol Med. 2024; 210: 108-119.
- Roininen SM, Cheetham M, Mueller BU, Battegay E. Unmet challenges in treating hypertension in patients with borderline personality disorder: A systematic review. Medicine (Baltimore). 2019; 98: e17101.
- Anderson G. Depression Pathophysiology: Astrocyte MitochondrialMelatonergic Pathway as Crucial Hub. Int J Mol Sci. 2022; 24: 350.
- Anderson G. Neuronal-immune interactions in mediating stress effects in the etiology and course of schizophrenia: role of the amygdala in developmental co-ordination. Med Hypotheses. 2011; 76: 54-60.
- Cui M, Aras R, Christian WV, Rappold PM, Hatwar M, Panza J, et al. The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc Natl Acad Sci U S A. 2009; 106: 8043-8048.
- Zárate SC, Traetta ME, Codagnone MG, Seilicovich A, Reinés AG. Humanin, a Mitochondrial-Derived Peptide Released by Astrocytes, Prevents Synapse Loss in Hippocampal Neurons. Front Aging Neurosci. 2019; 11: 123.
- Morris DL, Johnson S, Bleck CKE, Lee DY, Tjandra N. Humanin selectively prevents the activation of pro-apoptotic protein BID by sequestering it into fibers. J Biol Chem. 2020; 295: 18226-18238.
- Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003; 423: 456-461.
- Yang X, Zhang H, Wu J, Yin L, Yan LJ, Zhang C. Humanin Attenuates NMDA-Induced Excitotoxicity by Inhibiting ROS-dependent JNK/ p38 MAPK Pathway. Int J Mol Sci. 2018; 19: 2982.
- Jung JE, Sun G, Bautista Garrido J, Obertas L, Mobley AS, Ting SM, et al. The Mitochondria-Derived Peptide Humanin Improves Recovery from Intracerebral Hemorrhage: Implication of Mitochondria Transfer and Microglia Phenotype Change. J Neurosci. 2020; 40: 2154-2165.
- Markus RP, Fernandes PA, Kinker GS, da Silveira Cruz-Machado S, Marçola M. Immune-pineal axis - acute inflammatory responses coordinate melatonin synthesis by pinealocytes and phagocytes. Br J Pharmacol. 2018; 175: 3239-3250.
- Muxel SM, Pires-Lapa MA, Monteiro AW, Cecon E, Tamura EK, Floeter-Winter LM, et al. NF-κB drives the synthesis of melatonin in RAW 264.7 macrophages by inducing the transcription of the arylalkylamine-N-acetyltransferase (AA-NAT) gene. PLoS One. 2012; 7: e52010.
- Gao C, Liu M, Uzoechina J, Zhang Z. Oligodendrocyte lineage cells dysfunction in depression: early life stress, adolescent vulnerability and the emerging role of lipid metabolism. Transl Psychiatry. 2025: 22.
- Salahudeen T, Maalouf M, Elfadel IAM, Jelinek HF. Predicting depression severity using machine learning models: Insights from mitochondrial peptides and clinical factors. PLoS One. 2025; 20: e0320955.
- Miranda-Riestra A, Estrada-Reyes R, Constantino-Jonapa LA, Argueta J, Oikawa-Sala J, Reséndiz-Gachús MA, et al. Enhanced Modulation of CaMKII in Mouse Hippocampus by an Antidepressant-like Dose of Melatonin/Ketamine Combination. Cells. 2025; 14: 1187.
- Zhu W, Ding Z, Zhang Z, Wu X, Liu X, Zhang Y, et al. Enhancement of Oxytocin in the Medial Prefrontal Cortex Reverses Behavioral Deficits Induced by Repeated Ketamine Administration in Mice. Front Neurosci. 2021; 15: 723064.
- Anderson G. Natural Killer Cell Cytotoxicity: STAT3 Interactions with NF-κB Dimer Composition Modulate Mitochondrial Melatonergic Pathway: Tumor, and Viral Infection Treatment Implications. BIOCELL.
- Wang YH, Li JQ, Shi JF, Que JY, Liu JJ, Lappin JM, et al. Depression and anxiety in relation to cancer incidence and mortality: a systematic review and meta-analysis of cohort studies. Mol Psychiatry. 2020; 25: 1487-1499.
- Beurel E, Toups M, Nemeroff CB. The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron. 2020; 107: 234-256.
- Castle DJ. The complexities of the borderline patient: how much more complex when considering physical health Australas Psychiatry. 2019; 27: 552-555.
- Chin Fatt CR, Vasu S, Haque N, Ayvaci ER, Jha MK, Foster JA, et al. Cellular immune phenotype of major depressive disorder - findings from the EMBARC study. World J Biol Psychiatry. 2025; 26: 179-188.
- Wang S, Li M, Zhang C, Xu H, He J, Guo Y, et al. Discovery of 2,4-Disubstituted Pyrimidine Derivatives as Novel Kir4.1 Inhibitors with Rapid-Onset Antidepressant Effect. J Med Chem. 2026; 69: 1606-1627.
- Yovell Y, Bar G, Mashiah M, Baruch Y, Briskman I, Asherov J, et al. Ultra-Low-Dose Buprenorphine as a Time-Limited Treatment for Severe Suicidal Ideation: A Randomized Controlled Trial. Am J Psychiatry. 2016; 173: 491-498.
- Seo M, Anderson G. Gut-Amygdala Interactions in Autism Spectrum Disorders: Developmental Roles via regulating Mitochondria, Exosomes, Immunity and microRNAs. Curr Pharm Des. 2019; 25: 4344-4356.









































































































































{kind=link}