Affinity Analysis of CD16a- IgG Ligands Interactions
- 1. Department of Pathogen Biology, Hainan Medical University, P. R. China
Abstract
CD16a is the most key antibody Fc receptor expressed on human natural killer (NK) cells and is responsible to trigger NK cell-mediated ADCC. Therapeutic monoclonal antibodies (mAbs) require binding to CD16a for full effect and increasing the binding affinity. The point mutants were introduced into SCD1, CD16A mimetics, to enhance the binding affinity with IgG Fc fragment. Their three-dimensional structures were determined by homology modeling based on results previously determined by X-ray crystallography. The binding models were carried out via protein-protein docking tools. The best docking solutions were retained based on the docking scores and were used for structural assessment. Our results indicated that mutations in the CD16A receptor could strengthen binding affinity with IgG Fc fragment and might improve the interaction pattern of the neighboring residues. The stabilized mimetics of the small-molecule CD16A should serve as a promising target for bispecific antibodies that target the HIV-1 envelope protein and viral entry inhibitor.
Keywords
• Mutations
• CD16a mimetics
• Protein-protein interactions
• Modeling
CITATION
Zhang X, Lin J, Lin J, Huang L, He J, et al. (2024) Affinity Analysis of CD16a- IgG Ligands Interactions. Ann Virol Res 9(1): 1040
INTRODUCTION
Immunoglobulin G (IgG) antibodies play major roles in the control of bacterial and viral infections. Antibodies mediate antiviral effects by interfering with tumor cell growth or by activating antibody-dependent cell-mediated cytotoxicity (ADCC). The process of ADCC begins with recognition of an antigen expressed on the target cell surface. The Fc domain of antibodies is first bound by Fcγ receptors (FcγRs) expressed on immune effector cells, then triggers the release of cytotoxic granules towards the target cells or upregulates death receptors expression on the cell surface. ADCC is mainly mediated in humans by natural killer (NK) cells.
NK cells are considered to be the main mediators of ADCC both in physiological and therapeutic settings. CD16a/ FcγRIIIa is the primary receptor on NK cells that recognizes the IgGs-Fc segment and is responsible for triggering ADCC [1-3]. CD16a is a transmembrane receptor with a short C-ter cytoplasmic tail and two extracellular IgG-like structural domains [4]. It does not have any signaling components in its intracellular part. Therefore, it requires two immunoreceptor tyrosine-based activation motif (ITAM)-bearing signaling chains to transduce signals. Recent data show that the Fab fragment could be implicated in the FcγRIII/CD16a interaction [5,6]. Moreover, mutations in the Fab may modulate ADCC, highlighting the potential interaction of Fab-CD16a during IgG-CD16a binding [7].
Homology modeling is the most accurate of the computational structure prediction methods and has a vast range of applications in structure based drug development, analysis of mutations and binding mechanisms, identification of active sites, designing of novel ligands, protein–protein docking simulations etc20. Protein-protein interactions (PPIs) are universal to life and play a crucial role in cellular functions and biological processes in all organisms. Protein-protein interactions alter protein function by structural transformation [8-11].
The identification of protein interactions can lead to a better understanding of fundamental cellular regulation, infection mechanisms, the development of several medication drugs and treatment optimization. We hypothesized that the design of SCD1 with additional mutations might alter its affinity to cell receptor. In the present work, mutants were introduced into SCD1, CD16A mimetics. We attempted to predict the mutations impacted on protein-peptide binding affinity using computational methods. We performed computational studies and experiments to investigate the interactions between SCD1 and IgG. The online docking servers for protein-peptide interaction were utilized to predict the binding structures and identify the key residues between the SCD1 molecular and IgG. These findings revealed the polar and nonpolar intermolecular interactions contributing to SCD1/IgG binding stability, which provided an ideal target for viral entry inhibition and broad neutralization activity.
MATERIALS AND METHODS
CD16A sequences retrieval
The most basic data used in character analysis is sequence. Consequently, we started by searching the NCBI database for the CD16A sequence [12]. Ultimately, eight separate isoforms were obtained. The obtained sequences would be applied to additional research or analysis. The sequences were extracted from the database using the FASTA format.
Prediction of physicochemical properties
ExPASy proteomics tools (http://www.expasy.ch/tools/) provided the ProtParam online software, which could assess the most basic features including molecular weight, isoelectric point, and amino acid composition [13]. Here, we predicted the chemical composition, molecule weight, theoretical PI, and instability index of CD16A using ProtParam. In the meantime, we projected the charge and distribution of CD16A amino acids.
Sequences aligning and conservation analysis
The software MEGA 7 was used to align the recovered sequences [14], and Gblocks was used to assess the degree of conservation [15]. Sequence alignment, molecular evolution rate calculation, evolutionary hypothesis verification, and evolutionary tree inference may all be done with the useful program Mega [14]. Therefore, we aligned sequences using the Mega 7. The conservation sequence might be extracted via Gblocks [15].
Docking simulation assay
In order to identify the strength of interaction sites between CD16A and its antibody, we utilized ZDOCK online web server to predict two proteins binding spatial structure [16] and in virtue of pymol tool to visualize the complex and calculated the interaction sites with its strength [17]. ZDOCK online web server requires two input proteins, we chose CD16A PDB file 5D6D and CD16A Fab PDB file 7SEG downloaded in protein data bank (PDB web) [18-20]. We chose chains A, B, C, and D of the 5D6D, leaving chain C as the binding part. The results provided 10 best matches complexes; we picked the first 2 complexes to analyze strength of binding. The PDB files downloaded from ZDOCK web has been put to use for the next visual analyze. We utilized pymol tool to forecast the protein-protein interaction and calculated the hydrogen bond length.
RESULTS AND DISCUSSION
Physicochemical properties of CD16A
After obtaining our sequences, we used ProtParam online software to predict the physiochemical properties. We eventually the attained sequences 254 amino acids chain. The deficiency of amino acids determined three arrays of sequences distinctions. The 254-amino acid-chain consists of C1323H2016N246O379S8, whose molecule weight is 29089.13. The theoretical isoelectric points are 8.20. Besides, the instability indexes of all proteins are lower than 40, which signified that these proteins are stable. In the aspect of amino acid distribution, the most amino acid is leucine (Leu), accounts for 11% (28 in 254), 11.1% (28 in 253) and 12.5% (34 in 273). In 254-amino acid-chain, total number of negatively charged residues (Asp + Glu) are 25 while total number of positively charged residues (Arg + Lys) are 27. In 253-amino acid-chain, the number are 24 and 27 respectively. As for 273-amino acid-chain, the number of negatively charged residues add up to 28 and that of positively one decrease to 22.
Sequences aligning and conservation sites analysis
MEGA7 software was used to align all sequences (Figure 1), and Gblocks online software was used to analyze the conservation site among all 8 sequences (Figure 1) [14,15]. Utilization of MEGA 7 makes sequences orderly arranged. Conservative sites were provided by Gblocks and underlined in blue line. As a result, the conservation sequences were selected and exhibited in Table 1.
Table 1: Conservative sequences predicted by Gblock
|
Sequence number |
Position |
Sequences |
|
Conservation 1(C1) |
1-20 |
MWQLLLPTALLLLVSAGMRT |
|
Conservation 2(C2) |
22-192 |
DLPKAVVFLEPQWYRVLEKDSVTLKCQGAYSPED NSTQWFHNESLISSQASSYFIDAATVDDSGEYRC QTNLSTLSDPVQLEVHIGWLLLQAPRWVFKEED PIHLRCHSWKNTALHKVTYLQNGKGRKYFHHNS DFYIPKATLKDSGSYFCRGLFGSKNVSSETVNITITQ |
After aligning 8 arrays of sequences, we found that the redundancy caused the difference exists in three-type chains. Wiping of redundancy sequence and renumbering amino acid sequence, disappearing of E21 gave rise to distinction between the 254-amino acid-chain and the 253-amino acid-chain, which was exhibited in Figure 1,2.

Figure 1: Selected alignment blocks (underlined in blue).
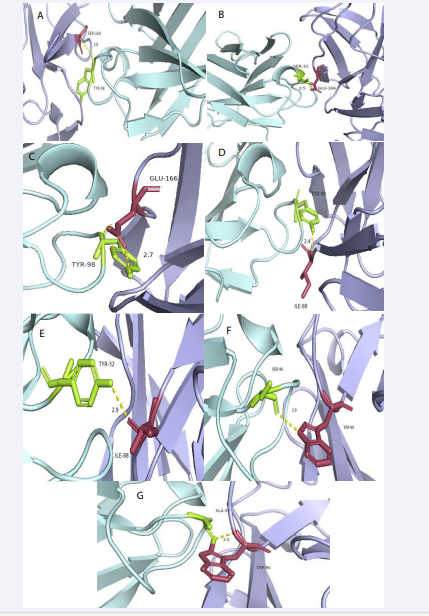
Figure 2: Visualized CD16A-IgG Fab complex. Nine pairs of interaction amino acids and CD16A/Fab is S151-Y54, S164-Y98, T167-S31, E166-S31, E166-Y98, I88-Y98, I88-Y32, W90-A97, W90-S96 respectively. (A1-I1) overall situation of amino acids interaction in whole complex; (A2-I2) nine groups of interaction sites between 2 proteins detail situation. (light blue for CD16A, limon for CD16A/Fab)
We subsequently used Gblocks to predict the conservation sites. Except the second sequence mentioned above, any other sequences possessed the same conservation sites which was tabulated later. We finally got 3 conservation sequences, C1, C2 and C3.
Docking
We determined the hydrogen bonding strength in complexes 1 and 2. There were nine locations in complex 1 where two proteins interacted with one another. In the Table 2,
Table 2: The interaction sites between CD16A and its Fab and strength of hydrogen bonds
|
No. |
CD16A amino |
Anti-CD16A antibody amino |
Strength of hydrogen bond |
|
1 |
S151 |
Y54 |
2.5 |
|
2 |
S164 |
Y98 |
3.0 |
|
3 |
T167 |
S31 |
3.4 |
|
4 |
E166 |
S31 |
2.5 |
|
5 |
E166 |
Y98 |
2.7 |
|
6 |
I88 |
Y98 |
2.4 |
|
7 |
I88 |
Y32 |
2.9 |
|
8 |
W90 |
A97 |
2.5 |
|
9 |
W90 |
S96 |
2.9 |
we have mentioned the contact sites and hydrogen bonding strength between the two proteins. We observed that I88, W90, and E166 in complex 1 might interact with several different amino acid sites, suggesting that these amino acid sites could be the binding sites that interact with antibodies. Thus, creating antibodies— even the antibody domains of bispecific antibodies—might be the goal.
CONCLUSION
We retrieved the CD16A sequences via NCBI for this investigation. Then, we examined the most basic characteristics of three different sequences: the 253 amino acid chain, the 254 amino acid chain, and the 273 amino acid chain. These characteristics included molecular composition, molecule weight, theoretical PI, and instability index. We also predicted the charge and distribution of CD16A amino acids in the interim. And the results showed that in the aspect of amino acid distribution, the most amino acid is leucine (Leu), accounts for 11% (28 in 254), 11.1% (28 in 253) and 12.5% (34 in 273). In 254-amino acid- chain, total number of negatively charged residues (Asp + Glu) are 25 while the total number of positively charged residues (Arg+ Lys) are 27. In 253-amino acid-chain, the number are 24 and 27 respectively. As for the 273-amino acid-chain, the number of negatively charged residues add up to 28 and that of positively one decrease to 22.
Next, we analyzed the conservation site among all 8 sequences. Fortunately, we got 3 conservation sequences, C1, C2 and C3. Then, we also predicted potential B-cell epitopes that can be potentially used as vaccine candidates and gave the sequence of 3 common B-cell antigenic epitopes in Conservation 1 (C1). When recognizing linear b-cell epitopes, antibodies can recognize denatured antigens. Loss of recognition for conformational B-cell epitopes would occur after antigen denaturing. Accounting for about a percentage of 90, B-cell epitopes are conformational. As a matter of fact, however, linear B-cell epitopes simply existed in minorities of native antigens.
Then, we estimated how accessible the surface would be. The consensus sequences with the chosen B-cell epitopes are present in two of the anticipated surface-accessible peptides, as we discovered. These two surface accessible peptides were MRTEDL and QWYRVLEK. It is a pity that the common sequences of C2 and C3 between their surface-accessible pepitides were not found. Finally, we forecasted the protein-protein interaction and calculated the hydrogen bond length. We found that I88, W90, and E166 could interact with multiple other amino acids, which indicated that these sites of amino acids could be the binding sites that interact with antibodies. Therefore, it could be the aim of designing antibodies, even antibody domains of bispecific antibodies.
CD16A is a potent cytotoxicity receptor on human natural killer (NK) cells, which can be exploited by therapeutic bispecific antibodies. Bioinformatics is based on mathematical and computer methods that could predict function and some other useful information about a protein with large quantities of information and a strong purpose. In this study, we used a combination methods of Expasy ProtParam tool, MEGA 7, Gblocks, BepiPred 2.0, IEBD?Karplus and Schulz (KS), Parker, and so on to predict the character of surface of CD16A. The features of CD16A, b cell epitopes, and the interaction between CD16A and anti-CD16A Fab were all investigated and anticipated in this work, which may have implications for the future development of peptide medications and vaccines.
ACKNOWLEDGEMENTS
This study was supported by research grants, Program for Hainan Provincial Key Research and Development Program (Grant No: ZDYF2022SHFZ077) and High-Level Talents of Hainan Province (Grant No: 2019RC213).
REFERENCES
- Thomas AS, Coote C, Moreau Y, Isaac JE, Ewing AC, Kourtis AP, et al. Antibody-dependent cellular cytotoxicity (ADCC) responses along with ADCC susceptibility influence HIV-1 mother to child transmission. JCI Insight. 2022.
- Monteiro MF, Papaserafeim M, Réal A, Yung GLP, Seebach JD. Anti- CD20 rituximab IgG1, IgG3, and IgG4 but not IgG2 subclass trigger Ca mobilization and cytotoxicity in human NK cells. J Leukoc Biol. 2020; 108: 1409-1423.
- Dhande JR, Bagul RD, Thakar MR. HIV-gp140-Specific Antibodies Generated From Indian Long-Term Non-Progressors Mediate Potent ADCC Activity and Effectively Lyse Reactivated HIV Reservoir. Front Immunol. 2022; 13: 844610.
- Woof JM, Burton DR. Human Antibody-Fc Receptor Interactions Illuminated by Crystal Structures. Nat Rev Immunol. 2004; 4: 89-99.
- Yogo R, Yamaguchi Y, Watanabe H, Yagi H, Satoh T, Nakanishi M, et al. The Fab Portion of Immunoglobulin G Contributes to its Binding to Fcγ Receptor III. Sci Rep. 2019; 9: 11957.
- Shi L, Liu T, Gross ML, Huang Y. Recognition of Human IgG1 by Fcγ Receptors: Structural Insights From Hydrogen-Deuterium Exchange and Fast Photochemical Oxidation of Proteins Coupled With Mass Spectrometry. Biochemistry. 2019; 58: 1074-1080.
- Sun Y, Izadi S, Callahan M, Deperalta G, Wecksler AT. Antibody- Receptor Interactions Mediate Antibody-Dependent Cellular Cytotoxicity. J Biol Chem. 2021; 297: 100826.
- Wippel HH, Chavez JD, Tang X, Bruce JE. Quantitative interactome analysis with chemical cross-linking and mass spectrometry. Curr Opin Chem Biol. 2022; 66: 102076.
- Musolino A, Gradishar W J, Rugo H S, Nordstrom JL, Rock EP, Arnaldez F, et al. Role of Fcγ receptors in HER2-targeted breast cancer therapy. J Immunother Cancer. 2022; 10: e003171.
- Wingert S, Reusch U, Knackmuss S, Kluge M, Damrat M, Pahl J, et al. Preclinical evaluation of AFM24, a novel CD16A-specific innate immune cell engager targeting EGFR-positive tumors. MAbs. 2021; 13: 1950264.
- Dormitzer PR, Grandi G, Rappuoli R. Structural vaccinology starts to deliver. Nat Rev Microbiol. 2012; 10: 807-813.
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Mizrachi IK, Lipman DJ,Ostell J, et al. GenBank. Nucleic Acids Res. 2009; 37: D26-D31.
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, William KL, Appel RD, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999; 112: 531-552.
- Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol. 2016; 33: 1870-1874.
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008; 36: W465-W469.
- Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014; 30: 1771-1773.
- Seeliger D, De Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010; 24: 417- 422.
- Ahmed AA, Keremane SR, Vielmetter J, Bjorkman PJ. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcγRIIIa. J Struct Biol. 2016; 194: 78-89.
- Kakiuchi-Kiyota S, Ross T, Wallweber HA, Kiefer JR, Schutten MM, Adedeji AO, et al. A BCMA/CD16A bispecific innate cell engager for the treatment of multiple myeloma. Leukemia. 2022; 36: 1006-1014.
- Berman HM ,Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000; 28: 235-242.









































































































































{kind=link}