General Overview on Recent Advance in Light-Induced Cu-I/II Photoredox Catalysis without Metal Co-Catalyst
- 1. University of Connecticut, India
- 2. Veer Narmad South Gujarat University, India
- 3. Goverment Science College, India
Abstract
The synergistic fusion of photoredox catalysis and transition-metal catalysis denominated metallaphotoredox reaction. Over the past decades, the merging of metal catalysis and photoredox catalysis has surfaced as a diverse and powerful technique in the production of challenging molecular synthesis. The vast dominance of photoredox concepts exploits heavy, precious and expensive metals. Iridium or ruthenium-based complexes obey single-electron reductants or oxidants in their photoexcited states. Thus, copper-based photocatalysts have rapidly sprung up in photochemistry due to their non-toxicity, abundance, low price, and novel properties, including inner coordination sphere mechanisms. Nevertheless, they exhibit tunable redox characteristics in their excited states, allowing stereoinduction accommodating flexible ligand architecture. Multiple accessible oxidation states are being accomplished for facile reductive elimination, oxidative addition, rapid radical capture and C−C/ C−N/ C−O/ C−S/ C−X cross-coupling reactions. We notice that there are plenty of research done on the concept of synergistic fusion of photo-redox and copper catalysis. Specifically, on this account we describe the latest advancements in light-mediated merged copper-photoredox catalysis as initiation in organic transformations, focusing on substrate scope, limitations, mechanistic paradigms, and various applications. This review covers the literature from 2018 to 2024.
Graphical Abstract

KEYWORDS
- Metallaphotoredox
- Photocatalysis
- Visible-Light
- Copper-Photoredox
- The Dual Catalytic System
CITATION
Viradiya RH, Choudhary A, Morja M, Chikhlia KH (2024) General Overview on Recent Advance in Light-Induced Cu-I/II Photoredox Catalysis without Metal Co-Catalyst. Chem Eng Process Tech 9(3): 1095.
ABBREVIATIONS
BINAP: (2,2′-Bis(Diphenylphosphino)-1,1′-Binaphthyl); Dtbbpy: 4,4’-Di-Tert-Butyl-2,2’-Bipyridyl; Bpin: Bis (Pinacolato) Diboron; TPA: Terephthalic Acid; Pypzs: (5-(4-Fluorosulfonyl) Amino-3-(2pyridyl)Pyrazole); Tpy: Terpyridine; Dap: 2,6-Diaminopurine; Dmbp: (6,6-Di-Methyl-2,2-Bipyridine); Ppy: 2-Phenylpyridine; PCN: Polymeric Carbon Nitrides; Bpy: 2,2′-Bipyridine; 1,10-Phen: 1,10-Phenanthroline; Acac: Acetylacetonate; TMS: Trimethylsilyl; Dfppy: (2-(2,4-Difluoro phenyl)Pyridine); TMSNCS: Trimethylsilyl Isothiocyanate; DIPEA: N,N-Diisopropylethylamine
INTRODUCTION
In a few decades, tremendous development is made in molecular synthesis, which led to a more efficient, greener and substantial advancement in organic chemistry. More than three centuries ago, researchers used transition metal complexes as catalysts for organic processes. During this period, study into transition metal catalysts has been developed and progressed in astonishing ways [1]. Transition metal catalysis has a significant impact on both science and society. Three Nobel prizes have been given out in recognition of this impact: in 2001[2], Knowles, Noyori, and Sharpless for stereoselective catalysis; in 2005 [3], Chauvin, Grubbs, and Schrock for olefin metathesis; and in 2010 [4], Heck, Negishi and Suzuki, for palladium- catalyzed cross-coupling. Although there have been significant improvements in metal-catalyzed cross-coupling reactions [5]. There are still notable limitations to this strategy, such as the pre- functionalization of starting materials and stoichiometric waste formation [6].
However, photoredox catalysis has lately come into prominence as a robust method for the SET (single electron transfer) process under very benign circumstances [7]. These techniques are valuable as a consequence of affordability, environmental friendliness and operational simplicity [8]. Due to the unfeasibility and limitations of the single photoredox catalytic system, the dual-catalytic system combining photocatalysis with transition metal enables the extension of an application under very mild circumstances [9]. Photoredox catalysis operated by visible-light has recently emerged as a very powerful, safer, non-polluting, renewable and user-friendly alternative [10]. In comparison to traditional approaches, the synergistic combination of photo-redox and transition metal catalysis harnesses light energy. Also, it has been proven as an ecologically benign and cost-effective solution for facilitating difficult C−C/C−N/ C−O/ C−S/ C−X bond formations. These dual systems additionally benefit from the high redox potentials of the photocatalysts, which permit control over the oxidation state of the transition metal catalyst. Hence allowing the formation of novel reactive pathways without the need for super- stoichiometric oxidants or reductants [11]. Photoredox-initiated reactions catalyzed by transition metal complexes or organic dyes (organic photoredox catalysts) have brought a significant revolution in organic synthesis [12]. Unfortunately, organic dye photocatalysts have weak photostability, making them less useful and less recyclable. To be an efficient photocatalyst, a molecule must have photostability, a long excited-state triplet with a lifetime (10-7-10-5 s), strong absorption in the visible region, high triplet yield, high extinction coefficient, and high reduction or oxidation potentials to transfer electrons to substrates [13]. It is essential to emphasize that this paradigm can bridge the gap between visible-light photoredox catalysis and metal catalysis [14], using elements such as rhodium [15], iridium [16], nickel [17], palladium [18], gold [19], and copper [20].
However, first-row transition metal-based photocatalysts, which have low toxicity, great abundance and cheap price are the best choice for avoiding these issues [21]. Especially, copper- based complexes [22], are becoming more desirable for visible- light-mediated reactions because of their inexpensive cost, availability, intrinsic characteristics, low toxicity and flexible catalytic systems. This synergistic reaction can be separated into two main categories. (A) Light-initiated copper catalysis. In this situation, several copper complexes are directly used as photocatalysts to drive the conversions, with no need for an external photosensitizer. (B) Cooperative copper/photoredox dual catalysis. In such a scenario, photosensitization of a conventional photocatalyst [23], starts the reaction by redox or atom transfer [24].
This review covers the relatively recent and significant advances that have been made using copper as photoredox catalysis in presence of visible light in organic synthesis. Here, we have highlighted the most recent significant advances made by the use of copper-based photocatalysts for organic synthesis. In this respect, most of the current reactions have been focused on C(sp3)–C(sp3), C(sp3)–C(sp2), C(sp3)–C(sp), C(sp2)–C(sp2), C(sp2)–C(sp), C(sp)– C(sp) and C-X (X=N,O,S,Cl,Br,I) bond formation.
LIGHT-INITIATED COPPER CATALYSIS
Carbon−Carbon Bond Formation
Catalytic C (SP3)-C(SP3) Bond Formation: C(sp3)—C(sp3) bond formation is difficult. The radical mechanism has always been shown to be an efficient method of achieving limitations. In 2018, moisture and air-stable dual copper-photoredox catalyzed three-component couplings protocol has been magnificently employed for the efficient assembly of trifluoromethyl thioethers using alkyl halides, olefins, and trifluoromethylthiolate as coupling partner by Peters and coworkers [Scheme 1] [25].
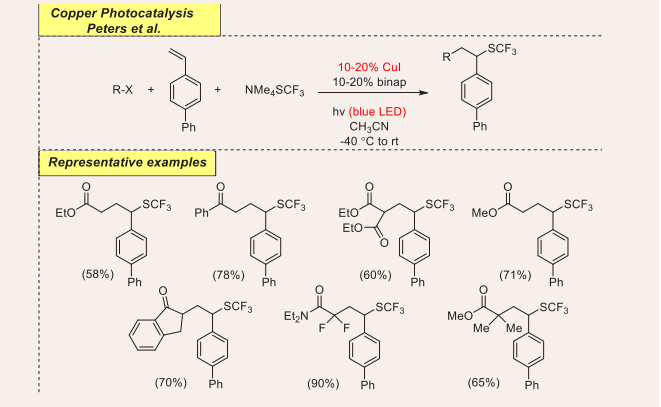
Scheme 1: Visible-light mediated copper-photoredox catalyzed cross-coupling reaction.
The initial reaction was carried out using olefins and an array of alkyl halides along with trifluoromethylthiolate as typical substrates. Optimization was carried out using 10-20% CuI as a photoredox catalyst, 10-20% Binap in methyl cyanide as the solvent for 24 h under blue LEDs irradiation. It is noted that various CuI and CuII sources also function as effective catalysts and provide similar consequences. However, without light, a CuI catalyst, and Binap, the transformation occurs with very low yields. Terminal olefins holding carbonyl, aryl or alkyl appear with moderate to a good yield of the corresponding product. In this coupling reaction, estrone acquires moiety and successfully provides a higher yield. A gram-scale synthesis of this procedure affords 84% yield of the aimed product, disclosing the potential of this synthetic approach. Yield is reduced when bromo carbonyl substrate is used as an electrophile.
Based on previous literature reports and these control experiments, a believable mechanism for this three-component protocol is projected as shown in [Scheme 2].
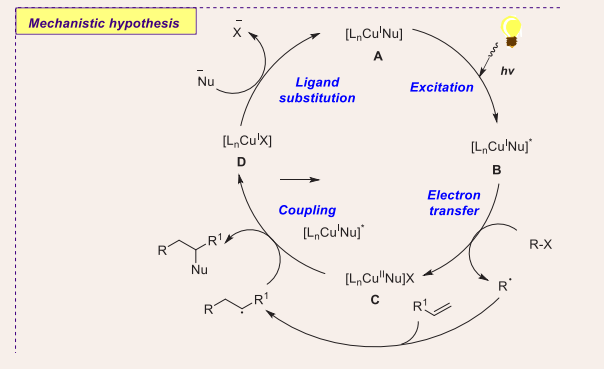
Scheme 2: Plausible mechanistic pathway of Cu-based photocatalytic coupling of alkyl Halides, olefins, and trifluoromethylthiolate.
Initially, copper catalyst [Ln(binap)CuI(SCF3)] A is photoexcited by blue LEDs to produce excited [Ln(binap)-CuI(SCF3)]* complex B, which is then involved in SET with an alkyl halide to furnish [Ln(binap) CuII(SCF3)] intermediate C with an alkyl radical. The addition of previously generated radical with alkene results in a new alkyl radical, which further reacted with the CuII radical to render the desired coupling product and generate halogen-containing CuI complex D. Finally, trifluoromethylthiolate completes the cycle by exchanging ligands with halogen-containing CuI complex D, resulting in complex [Ln(binap) CuI (SCF3)] A.
At the end of 2018, a straightforward strategy to access biologically fascinating 1,1-diarylmethane-containing alkylnitriles via radical cross-coupling (RCC) of boronic acids, styrene and oxime esters under visible-light and the copper catalyst was introduced by Xiao group [Scheme 3] [26].
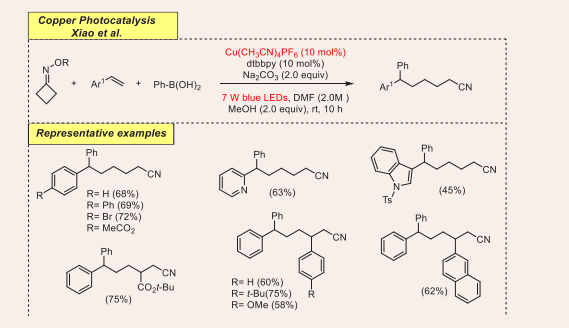
Scheme 3: Visible-light mediated copper-photoredox catalyzed radical cross-coupling reaction.
The initial transformation has been achieved by using cyclobutanone oxime ester, phenyl boronic acid and 2-vinylnaphthalene.
The optimized conditions for this reaction are as follows: 10 mol% of Cu(CH CN) PF as catalyst, 10 mol%) of 4,4’-di-tert butyl-2,2’-bipyridyl (dtbbpy) as ligand and K2CO3 as a base in dimethyl formamide under 7 W blue LEDs irradiation at room temperature. They found that the presence of the copper catalyst, base and light irradiation is essential for getting desired coupling products. Aryl boronic acids containing both electron-efficient and electron-deficient substituents at the para position of benzene ring react quite smoothly and well dispensing average to good yields. Remarkably, heteroaryl-substituents on alkenes tolerated nicely with excellent yield. O-acyl oxime esters bearing substituted phenyl, ether, naphthyl and ester functionalities were scrutinized, despite the electronic nature, and moderate to good yields of the aimed products were observed. There are several notable advantages to this reaction which include its wide substrate scope, straightforward technique, inexpensive copper catalyst and oxidant-free approach.
To get insight into the mechanistic features, the authors have performed radical trapping experiments and radical clock experiments [Scheme 4].
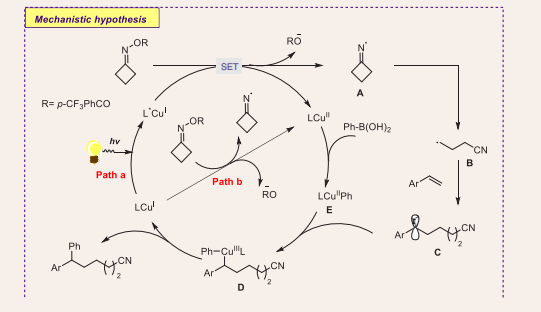
Scheme 4: Plausible mechanistic pathway of Cu-based photocatalytic radical cross-coupling of boronic acids, styrene and oxime esters.
Initially, iminyl radical A can be afforded via single electron transfer reduction of oxime ester through the excited state of the *CuII complex, which is allowed in accessing the CuII complex (Path a). Next, cyanoalkyl radical B may be generated through β-cleavage of radical A, which is captured by 2-vinylnaphthalene to dispense a new and stable radical C. Later, alkyl radical C is tackled by CuII complex E, which is readily formed via a transmetalation reaction between copper complex and phenyl boronic acid, to CuIII intermediate D. Lastly, the desired coupling product is obtained by reductive elimination of CuIII species D, and the CuI catalyst is returned for another cycle.
After a year, the same research group developed a simple strategy for the formation of regioselective C(sp3)—C(sp3) bonds, resulting in the desired cross-coupled product. In this reaction cyanoalkyl-containing propargylic works as a precursor via appropriate coupling partners such as terminal alkynes, alkenes, and cycloketone oxime esters [Scheme 5] [27].
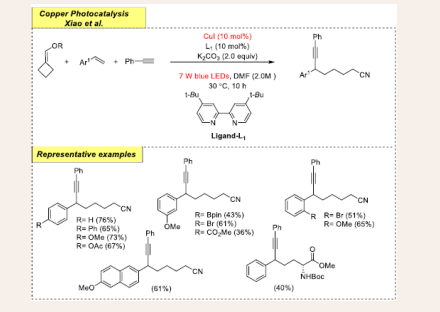
Scheme 5: Visible-light mediated copper-photoredox catalyzed radical cross-coupling reaction.
They accomplished the initial reaction by using 2-vinylnaphthalene, cyclobutanone oxime ester and phenylacetylene as benchmark substrates. Optimization studies reveal the use of CuI (10 mol %) as catalyst, 4,4′- dimethoxy-2,2′-bipyridine (L1) as a ligand (20 mol %), and K2CO3 (0.6 mmol) as a base in solvent DMF (2.0 mL) at 30°C under7 W blue LEDs irradiation for 10h. The radical trapping stage is made easier by using a super-stoichiometric quantity of base. Even after an extended reaction period, the ultimate yield was drastically reduced without visible-light irradiation for 10h. The substrate scope of the procedure was fairly reasonable. Under an optimum reaction medium, it is observed that the electron- poor (e.g., CO2Me, Br, Bpin) and the electron-rich (e.g., OAc, OMe, Ph) functionalities at the para position synthesize products with good to average yields. Moreover, the other alkene substrates containing estrone, amino acid, gibberellic-acid, and febuxostat scaffolds produce the coupling products in modest to good yields. Disappointingly, the absence of oxime ester renders detrimental outcomes for the desired coupling product. Mild conditions, a wide substrate range, and a high tolerance for functional groups make this technique ideal for accessing structurally varied propargylic compounds. Mechanistic investigations reveal that benzylic radical is an essential intermediate, which is produced with the addition of a cyanoalkyl radical to styrene and involves a SET mechanism.
In 2019, Zhao and co-workers established a copper- photo-catalyzed reaction for the preparation of biologically and pharmaceutically important tetrazole-decorated triphenylamine based on MOF (metal–organic framework) {TBA[Cu2(TPA)]}·CH3CN via oxidative C(sp3)−C(sp3) coupling of phenyl tetrahydroisoquinoline and nitromethane [Scheme 6][28].
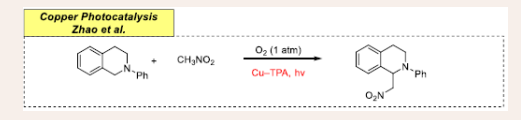
Scheme 6: Visible-light mediated copper-photoredox catalyzed oxidative coupling.
N-phenyl-tetrahydroisoquinoline (0.2 mmol), reacted with nitromethane (2.0 mL) in the presence of a Cu–TPA organo-photocatalyst (3.0 mol%), under the irradiation of 26 W fluorescent lamp using molecular oxygen as an oxidizing agent. These heterogeneous photocatalytic frameworks provide an eco- friendly path for broadening the scope of C-C bond formation. The result suggests the successful implementation of tetrazole- containing triphenylamine-based MOF design, with high reaction efficiency.
In a continuous interest in metallaphotoredox catalysis, Gong et al., improvised a novel approach for the emergence of acyclic imine derivatives. The reaction proceeded via organophotoredox and chiral bisoxazoline copper catalyst-mediated enantioselective α-aminoalkylation from N-acylhydrazones and α-silylamine moieties [Scheme 7][29].
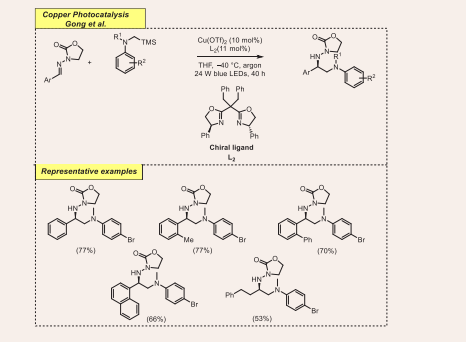
Scheme 7: Visible-light mediated copper-photoredox catalyzed α-aminoalkylation.
In particular, this transformation is facilitated by premixed Cu(OTf)2 salt, chiral ligand L2 (10 mol%), in the presence of THF under argon at standard temperature using 24W blue LEDs radiation. Here mentioned combined approach tolerates a wide category of electron-rich and electron- poor groups bearing N-acylhydrazones, whereas alkylsubstituted aldimine dispenses deleterious outcomes. Regarding the α-aminoalkyl radical precursors, tertiary α-silylamines and electron-withdrawing α-silylamines were well tolerated in the reaction.
A comprehensive mechanism was predicted by the Golg group, shown in [Scheme 8].
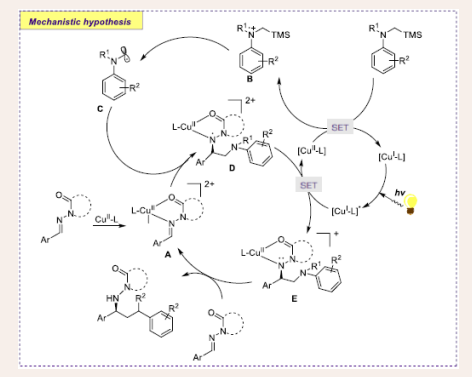
Scheme 8: Plausible mechanistic pathway of Cu-based photocatalytic α-aminoalkylation of acyclic imine derivatives.
Initially, [L-CuII] goes through ligand exchange with N-acylhydrazone species furnishing intermediate A. Consequently, SET between [L-CuII] and α-silylamine dispensing radical cation B and a [L-CuI] species. Later, TMS cation readily dissociates to α-aminoalkyl radical C, followed by reduction of [L-CuI]* rendering monocationic complex E, which on protonation and ligand exchange furnishes chiral diamine product.
Copper-based photocatalyzed pyrazole-pyridine ligand incorporated a sulfonamide moiety-promoted, proton-coupled electron transfers (PCETs) process for reductive Pinacol type couplings of ketone or aldehyde derivatives have been successfully demonstrated by Collins et al., in 2019 [Scheme 9] [30].
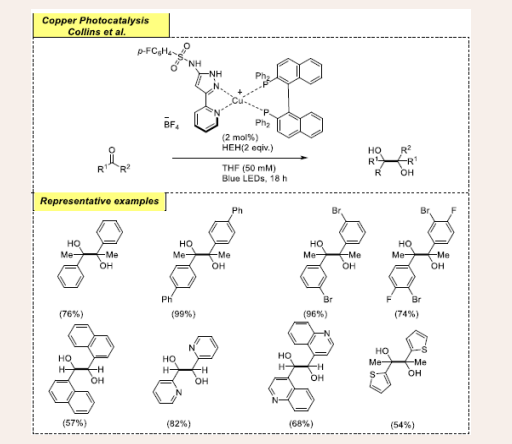
Scheme 9: Visible-light mediated copper-photoredox catalyzed reductive pinacol-type coupling reactions.
The reaction conditions are optimized by variations of the Cu sources, ligands, additives, solvent, and temperature. The promising optimized reaction conditions involved Cu(pypzs) (BINAP)BF4 as a catalyst, diphenyl phosphoric acid (dppa) as an external acid and Hantzsch ester (HEH) as a hydrogen-atom donor in THF under blue LEDs. Remarkably, the reaction stopped completely, and no desired product was obtained in the absence of dppa. Pyrazole-pyridine ligand incorporates a sulfonamide moiety, that functions as an intramolecular hydrogen-bond donor for a photochemical PCET process. Next, the substrate scope of this pinacol-type coupling reaction was examined using both aldehydes and ketones to afford diols. Notably, halo-substituted prototypical substrates furnish moderate to excellent yields. Emission quenching studies predicted that the reaction would involve a SET radical mechanism.
In 2020, Larionov and colleagues investigated the dual- photo-copper catalytic strategy for useful C(SP3)-C(SP3) bond installation using carboxylic acids as stable radical precursors and long-chain aldehyde using an acridine/copper catalytic system [Scheme 10] [31].
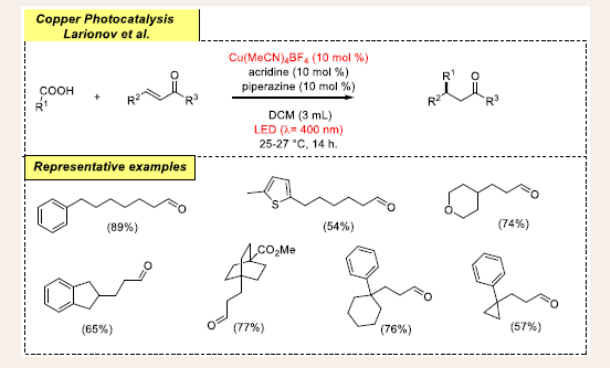
Scheme 10: Visible light mediated copper-photoredox catalyzed cross-coupling reaction.
This reaction is carried out in air at 25-27°C for 14 hours in the presence of 10% Cu(MeCN)4BF4, acridine (10 mol%), DCM (3 mL), piperazine (10 mol%), and LED (400 nm). This decarboxylative dual-catalytic conjugate addition reaction proceeds with lewis acids because the catalytic activity of copper is facilitated only due to lewis acidity. Carboxylic acids have a critical role in making decarboxylation simpler by activating reactive esters, which are typically necessary for decarboxylation. Remarkably, this transformation allows access to various copper cocatalysts by extension, to acridine. It reveals that Cu(MeCN)4BF4 catalyst is superior to CuCl, CuOTf and CuBr2. Pleasingly, this dual protocol is well tolerated with various functional groups such as chloro, fluoro, bromo, heteroaromatic groups, unprotected alcohols, etc. in the carboxylic acid derivatives, delivering structurally diverse products in moderate yields. The scope of the reaction is further expanded with ketones and aldehyde derivatives. Even though most of the parent substrates work well in normal conditions. Also, there are a few key things that can be changed to increase yields.
According to the author’s anticipated mechanism, presumably, acridinyl radical G is generated through the light-induced proton- coupled electron transfer (PCET) in acridine acid complex B. It can reduce CuI complex C, emerging Cu0 species D and acridinium G. A conjugate addition to a Michael acceptor may be performed on the alkyl radical that was most likely created during the acridine- catalyzed decarboxylation. After a cross-termination with the Cu0 complex D, the resultant-carbonyl radical E may produce the CuI enolate F. Both CuI complex C and acridine photocatalyst A is expected to regenerate after the following protonation of enolate F by acridinium G, ending both catalytic cycles. The acridinyl radical H is found to be an intermediate in the redox turnover of the acridine and copper catalytic cycles. This also shows that the CuI/Cu0 catalytic cycle works well [Scheme 11].
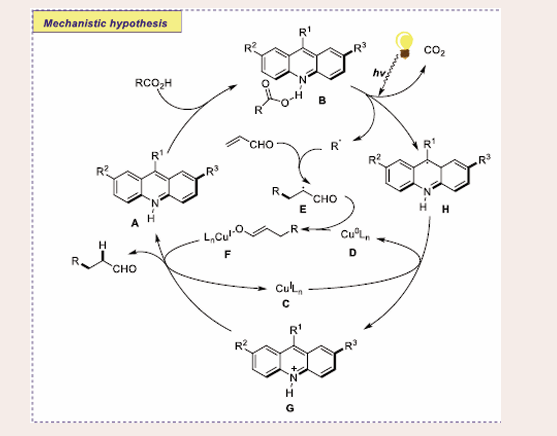
Scheme 11: Plausible mechanistic pathway of Cu-based photocatalytic α-aminoalkylation of acyclic Imine derivatives.
A straightforward process for oxy-monofluoromethylation of alkenes is presented, utilizing visible-light CuI photoredox catalysis discovered by Veliks. An iodine(III) reagent with monofluoroacetoxy ligands acts as an efficient source of the monofluoromethyl (CH2F) radical, allowing the step-efficient synthesis of γ-fluoro-acetates from various olefinic substrates under mild conditions [Scheme 12][32].
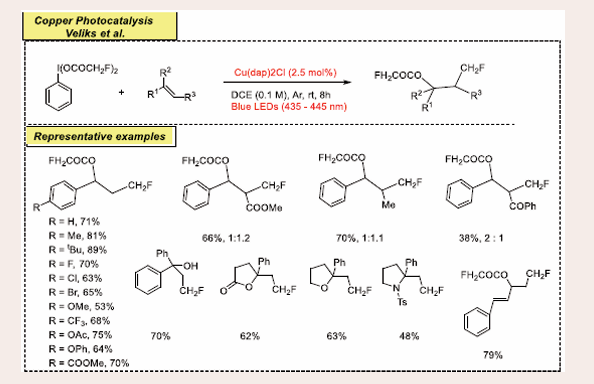
Scheme 12: Visible light mediated copper-photoredox catalyzed Oxy monofluoromethylation of Alkenes.
This method is also applicable to late-stage diversification of alkenes from complex molecules, amino acids, and the synthesis of fluoromethylated heterocycles. And reported a direct oxy-monofluoromethylation of a broad range of olefinic substrates, including biologically relevant molecules, by combining copper(I) photoredox catalysis with iodine(III) chemistry. Additionally, the utility of this method in synthesizing various CH2F-containing heterocycles is demonstrated.
Vinylarenes with various electron-donating and electron- withdrawing groups on their phenyl ring produced products in 53–89% yield. The protocol was successfully applied to α,β- unsaturated compounds and an internal alkene, yielding oxy- monofluoromethylation products with diastereoselectivities from 1:1 to 4:1. For 1,1-disubstituted alkenes, 1,1-diphenylethylene yielded monofluoromethylated alcohol in 70%. This protocol extended to an intramolecular version, yielding monofluoromethylated γ-lactam, tetrahydrofuran, and N-tosyl- pyrrolidine in moderate to good yields. A conjugated diene delivered the terminal C=C bond monofluoromethylesterification product with 79% yield.
Based on the EPR and control experiments, a plausible mechanistic pathway is proposed. The iodine(III) reagent undergoes heterolytic cleavage of one I–O bond, generating cationic species A. This species is reduced by excited *[Cu(dap)2]+, producing radical iodine intermediate B. Homolytic cleavage of the remaining I–O bond in B forms PhI and a fluoroacetoxyl radical, which decarboxylates to produce a monofluoromethyl radical (C) and releases CO2. Visible light may accelerate this decarboxylation step. The monofluoromethyl radical (C) then adds to the alkene, forming benzyl free radical D. The photoredox cycle is completed by a single electron transfer from D to [Cu(dap)2]2+, generating benzyl carbocation E, which is trapped by the fluoroacetate anion to yield the desired product [Scheme 13].
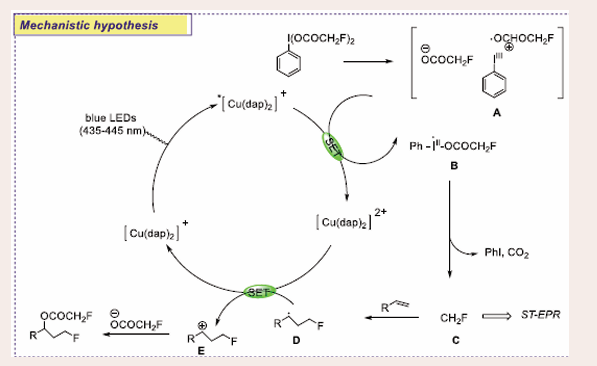
Scheme 13: Plausible mechanistic pathway of Cu-based photocatalytic Oxy-monofluoromethylation of Alkene
A new strategy for visible light-induced, copper-catalyzed three-component difluoroalkyl thiocyanidation of alkenes has been developed to synthesize a series of valuable difluorothiocyanate compounds by Hu [Scheme 14] [33].
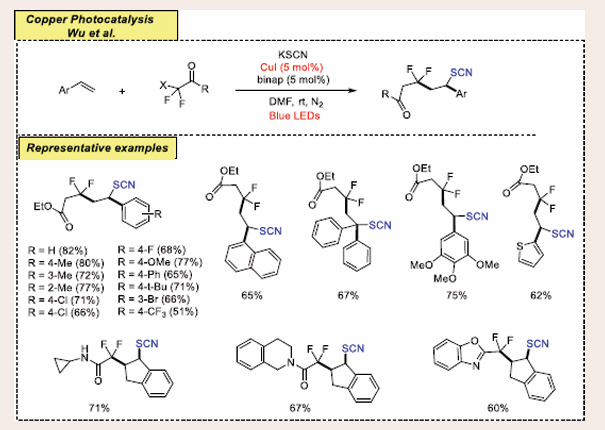
Scheme 14: Visible-Light-Induced Copper-Catalyzed Intermolecular Three-Component Difluoroalkyl Thiocyanidation of Alkenes.
This method can also be extended to perfluorothiocyanate compounds and applied to target molecules with drug or natural product skeletons. Mechanistic studies reveal that the copper complex serves a dual role, functioning as both the photoredox catalyst for electron transfer and the cross-coupling catalyst for C−SCN bond formation.
Aromatic alkenes with ortho-, meta-, or para-substituents, both electron-donating and electron-withdrawing, showed good reactivity under standard conditions, yielding the desired products in moderate to good yields. Unexpectedly, the reaction with 1,1-diphenylethylene produced an isothiocyanate product instead of the anticipated thiocyanate, likely due to steric hindrance influencing SCN capture at sulfur versus nitrogen. Additionally, 2-vinylthiophene delivered product in 62% yield. Additionally, also found that both cyclic and acyclic N,N- dialkyl amides were effective coupling partners, yielding the corresponding products in varying yields ranging from 60% to 71%. Plausible mechanism is same as above mention.
Catalytic C(SP3)-C(SP2) Bond Formation
In 2020, Pericas et al., introduced a visible-light-mediated copper-catalyzed stereodivergent formation of allylic amines and ethers through the decarboxylative hydroalkylation of alkynes and carboxylic acid derivatives [Scheme 15] [34].
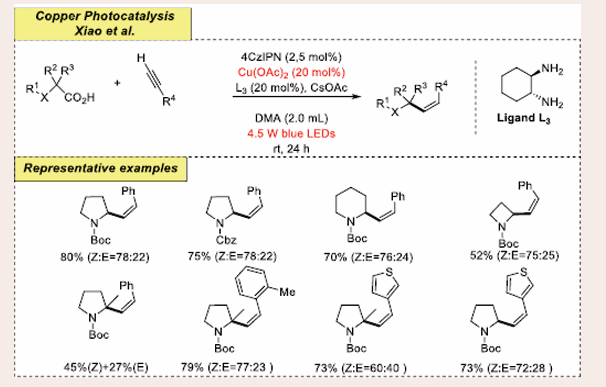
Scheme 15: Visible-light mediated copper-photoredox catalyzed decarboxylative hydroalkylation.
The primary reaction was executed using N-Boc Proline and phenylacetylene as the preliminary substrates. The protocol requires Cu(OAc)2 as a copper-catalyst, 4CzIPN as a photocatalyst3 a ligand system and CsOAc as the base, in DM at normal room temperature under blue LED. They were able to isolate the intended product in 82% yield with a Z:E ratio of 78:22 through the ideal parameters. In this protocol, various α-amino radicals (primary, secondary, tertiary) and secondary α-oxy radicals like other functional groups could couple with alkynes to furnish their corresponding hydroalkylated products in high yields via Z-selective manner. In addition, heteroaromatic alkynes and enyne also successfully participated in this transformation with E-selectivity. But interestingly, no hydroalkylated product is observed when non-conjugated alkynes (1-hexyne and tert-butyldimethylsilylacetylene) were employed in the current transformation.
Based on the deuterium atom abstraction investigation, the authors hypothesized that a radical mechanism was involved [Scheme 16].
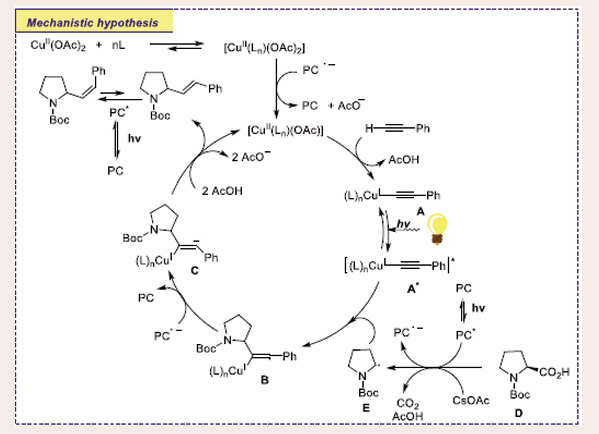
Scheme 16: Plausible mechanistic pathway of Cu-based photocatalytic decarboxylative hydroalkylation of alkynes and carboxylic acid derivatives.
The photocatalyst 4CzIPN attains its excited state after exposure to light and is then able to oxidize-amino and-oxy carboxylates as well as diminish the CuII complexes. Stern- Volmer studies show that [Cu(L3)2](OAc)2 has a slower reductive quenching of the catalyst than Boc-Pro-OCs. Complex A may be formed by using a base CsOAc to disproportion the CuII complexes or by using SET by PC. Alkyne moiety charge depletion via ligand-to-metal transfer (LMCT) could be determined by direct photoexcitation of A to A*, which would speed up the assault of radical E on the alkyne moiety. The vinyl radical B is formed as a consequence of this addition, and the vinyl anion C is formed as a result of PC being oxidized. The product may be synthesized by protonation of an anion C and proto-demetalation of the Cu-C bond.
The development of value-added transformations of naturally abundant amino acids has garnered significant interest in the synthetic chemistry field. While asymmetric decarboxylation reactions are well-established, the asymmetric deamination of amino acids remains challenging due to difficulties in C–N bond activation and potential incompatibility with chiral catalysts. Herein, Xiao at al., presented a photoinduced, copper- catalyzed asymmetric deaminative coupling of amino acids with arylboronic acids. This method enables the synthesis of a diverse range of valuable chiral phenylacetamides with high yields (42–85%) and excellent enantioselectivity (up to 97% ee) under mild, environmentally friendly conditions [Scheme 17] [35].
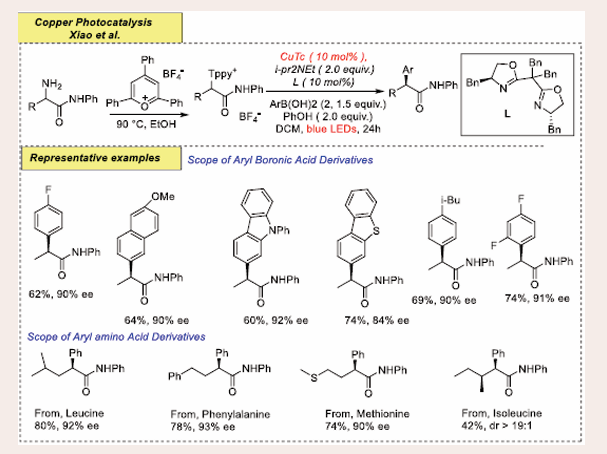
Scheme 17: Visible Light Induced Copper-Catalyzed Enantioselective Deaminative Arylation of Amino Acid Derivatives
Experimental studies and theoretical calculations highlight the pivotal role of added phenols in enhancing both catalytic efficiency and enantioselectivity. As shown in Figure, various arylboronic acids were effective in this transformation, producing the corresponding chiral α-aryl N-phenylamide products with yields ranging from 52% to 85% and enantioselectivities of 84% to 97%. The reaction tolerated a range of electronic and substitution patterns on the benzene ring, resulting in moderate to good yields and high enantioselectivities (52–85% yields, up to 97% ee). Arylboronic acids with electron-deficient substituents, such as acetyl, ester, and sulfonate, resulted in lower yields but maintained high enantioselectivity. Arylboronic acids with multiple substituents also gave good results (74–77% yields, 91% ee). Fused aromatic and heteroaromatic boronic acids, including 6-MeO-2-naphthyl, carbazolyl, and dibenzothiophyl- substituted acids, were converted with yields of 60–74% and enantioselectivities of 84–92%. The reduced enantioselectivity of product 3p was likely due to steric hindrance from an ortho-substituent. Also further explored the scope of amino acid derivatives. Natural α-amino acids, such as leucine, phenylalanine, methionine, isoleucine, and glutamic acid, reacted well, yielding enantio-enriched amides in 42–80% yields and 90–93% ee. Unnatural amino acids, including norvaline and ornithine derivatives, also produced good results (75% yield, 92% ee; 74% yield, 84% ee). N-substituted amides also well tolerated, delivering product 48% yields with 91% ee.
Upon blue light irradiation, the CuI complex transitions to an excited state, facilitating a single electron transfer to the Katritzky salt, generating α-amide radicals. Subsequently, a phenoxide ligand coordinates with the CuII complex, enabling a transmetallation process with the arylboronic acid to form an aryl-Cu intermediate. This aryl-Cu species then intercepts the α-amide radicals, resulting in the release of the deaminative arylation product. This mechanism highlights the sequential electron transfer, ligand coordination, and radical capture, leading to the formation of the desired product [Scheme 18].
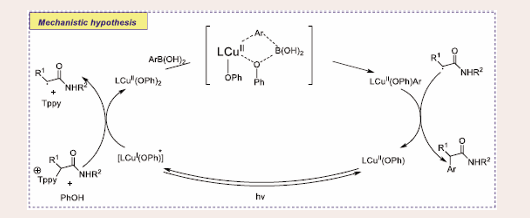
Scheme 18: Plausible mechanistic pathway of Visible Light Induced Copper-Catalyzed Enantioselective Deaminative Arylation of Amino Acid Derivatives.
Catalytic C(SP3)-C(SP) Bond Formation
In parallel to this study, Zhang and co-workers magnificently unfolded a facile and enantioselective light-induced copper- catalyzed direct alkynylation and arylation/alkylation approach of alkenes to afford valuable propargylic compounds [Scheme 19][36].
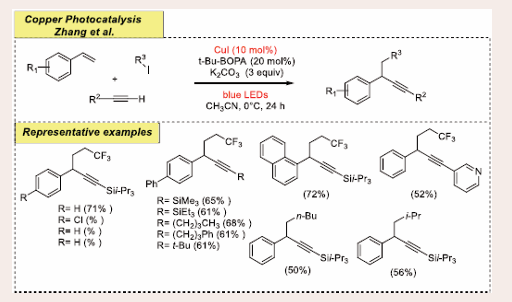
Scheme 19: Visible light mediated copper-photoredox catalyzed carbofunctionalization.
The optimized reaction conditions are as follows: 10 mol% of CuI as the photocatalyst, 20 mol% of t-Bu-BOPA as the ligand and 3 equiv of K2CO3 as a base in CH3CN at 0°C under blue LEDs. Monosubstituted alkenes, alkynes as well as aryl and alkyl iodides are subjected to photoredox couplings which utilize only a single optically active copper complex, resulting in valuable enantioselective propargylic compounds and the building block of pharmaceuticals. Substituting styrenes with various functional groups such as OCH3, Cl, Br, CH3 and t-Bu at the para position leads to difunctionalization of alkenes with haloalkanes in better yield. Also, changing t-Bu-BOPA with other ligands gives rise to diminished titled product generation. The successful capability of this technique was disclosed by various linear aliphatic alkynes, haloalkanes and both primary and secondary aliphatic iodides.
The mechanism of the direct difunctionalization is investigated via radical-trapping investigation using TEMPO and Stern−Volmer experiments [Scheme 20].
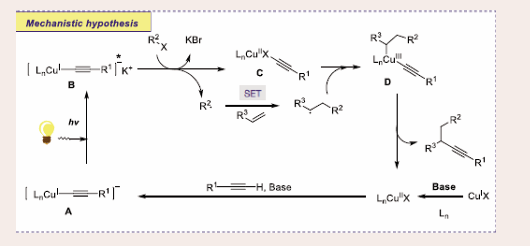
Scheme 20: Plausible mechanistic pathway for Cu-based Photocatalytic carbofunctionalization of alkenes.
In the basic medium, the excited CuI complex B is generated through photo-irradiation on CuI complex A, which furnishes alkyl radical and CuII complex C through SET. Next, the alkyl radical is captured by styrene to deliver a benzylic radical, which readily reacts with CuII complex C dispensing CuIII complex D. Finally, the desired product is created by reductive elimination of CuIII complex D.
In 2020, Shi et al., introduced photo initiated copper-catalyzed efficient and sustainable approach for the preparation of C(sp3) – Csp bond via cross-coupling reaction between alkynes and oxime esters [Scheme 21] [37].
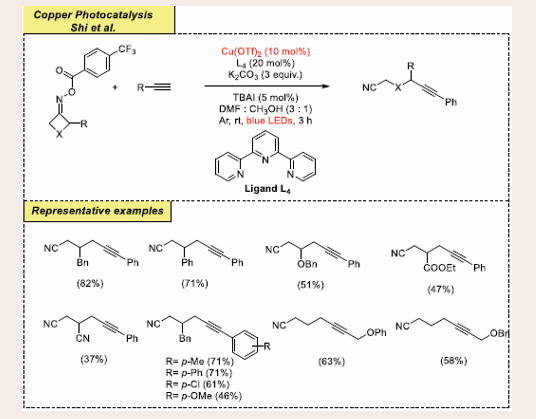
Scheme 21: Visible-light mediated copper-photoredox catalyzed cross-coupling reaction.
The initial optimized reaction is carried out with terminal alkyne; phenylacetylene and cyclobutanone O-(4-(trifluoromethyl)benzoyl)oxime as a model substrate. This reaction is carried out in presence of 10 mol% of Cu(OTf)2 as copper-catalyst, 20 mol% of ligand L4, 3 equiv of base K2CO3 and 5 mol% of TBAI in a 3 : 1 mixture of DMF: CH3OH, under blue LEDs and argon atmosphere, at room temperature for 3 hours. It has been observed that the presence of copper-catalyst, base, ligand and visible-light is mandatory for getting good yields of the target product. This protocol exhibited a good substrate scope, decent reactivity and reverence to both the coupling partners. Cyclobutanone oximes containing cyano and ester substitution on four-member ring substrates are less effective and produce desired 37% and 47% isolated yield respectively due to alcoholysis. However, OBn, Ph and Bn containing substrates give the wanted products moderate to good yields in these protocols. Also, it has been reported that a wide range of substrate scopes of phenylacetylene substituted by multiple groups including −OMe,−Me, −Cl, and −Br at ortho, meta and para-positions facilitate the reaction.
Based on previous literature reports and control experiments, such as radical inhibitor experiments and competitive experiments, a plausible mechanism for this protocol is projected as shown in [Scheme 22].
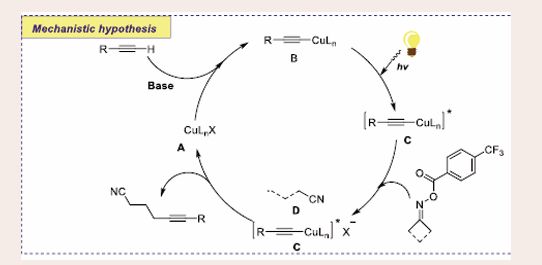
Scheme 22: Plausible mechanistic pathway for Cu-based photocatalytic cross-coupling reaction of alkynes and oxime esters.
Initially, excited copper complex B is dispensing through photo-irradiation on copper complex A, Due to the higher reductive potential of excited copper complex B readily involved in reductive single electron transfer to furnish γ-cyanoalkyl radical D and copper complex C. Simultaneous, radical D can easily be trapped by complex C to release desire product alkynyl nitriles and get back copper catalyst A. Elimination of extra photoredox catalyst and oxidants, low catalyst loading and operational simplicity make this transformation more auspicious.
In the same year, Pan and co-workers further introduced a technique for the construction of the C(sp3) –Csp bond via decarboxylative alkynylation reaction of carboxylic acids especially RAEs (Redox-active esters) of carboxylic acids [Scheme 23] [38].
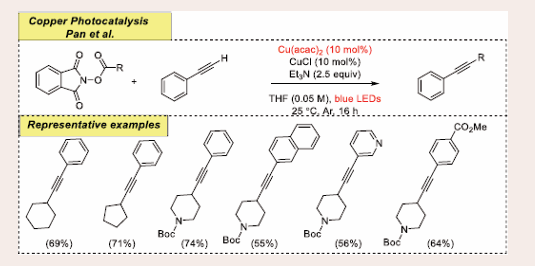
Scheme 23: Visible-light mediated copper-photoredox catalyzed decarboxylative alkynylation reaction.
Under the optimized conditions, 1.0 equiv of alkynes, 1.5 equiv of RAE as model substrates including the equivalent mixture of Cu(acac)2 and CuCl (10 mol%), and 2.5 equiv of base Et3N in THF under 90 W blue LEDs irradiation this transformation occurs smoothly. The reaction is shut down in absence of Et3N, CuCl, Cu(acac)2 and in darkness. It turns out that hetero-cycles and cyclic hydrocarbons containing carboxylic acids provide the expected product with a good yield. Interestingly, the optimized conditions are also suitable for other heterocycles such as benzothiaozyl, thiophene, indoyl, naphthyl, pyridyl and carbazoyl derivatives. Surprisingly, unprotected amines do not respond under standard conditions.
Based on DFT calculations and computational evidence plausible mechanism for this protocol is projected as shown in [Scheme 24].
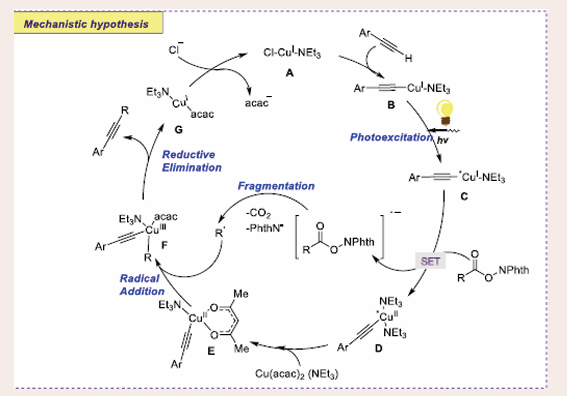
Scheme 24: Plausible mechanistic pathway for Cu-based Photocatalytic decarboxylative alkynylation reaction of alkynes and RAEs of carboxylic acids.
Initially, in the basic medium, CuI complex A is generated, which reacts with the alkyne to furnish CuI acetylide B, which on irradiation generated copper acetylide C. Through SET process with RAEs gives a radical anion of the redox-active esters and CuII acetylide complex D. Next, decarboxylative fragmentation of complex D renders an alkyl radical. Ligand exchange between Cu(acac) (NEt ) and CuII-acetylide complex D dispensing the copper complex E. The radical addition of the alkyl radical to copper complex E gives hypervalent CuIII complex F. Finally, the reductive elimination of CuIII complex F affords the desired cross-coupling product and G.
During this time, Wang, Liang, and Liu’s group have also developed a simple method for chiral C(sp3)-C(sp) bond formation via light-mediated copper-catalyzed decarboxylation with the use of appropriate execution of coupling partners takes place by using a racemic mixture of NHP (N-hydroxyphthalimide) esters of the carboxylic acid with terminal alkynes [Scheme 25] [39].
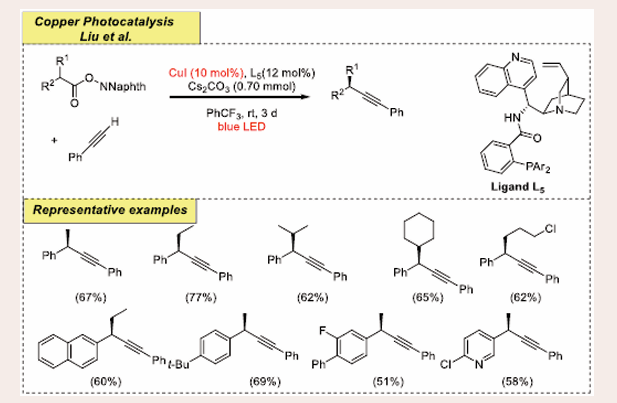
Scheme 25: Visible-light mediated copper-photoredox catalyzed decarboxylative alkynylation reaction.
They accomplished that the initial reaction occur by using 2-phenylpropanoic acid and phenylacetylene as benchmark substrates. Optimization studies reveal that the use of CuI (10 mol%) as a catalyst, ligand L5 (12 mol%), and Cs2CO3 (0.70 mmol) as the base in solvent PhCF3 (4.0 mL) under blue LEDs for 3 days at room temperature appears with the best results. It has also been observed that no reaction occurs without the use of ligands and light. Under optimum reaction medium, it is found that the electron-poor and the electron-rich functionalities both synthesize products with good to average yields with good regioselectivity. The NHP-type esters of anti-inflammatory medications such as Pranoprofen, Ibuprofen, Zaltoprofen and Flurbiprofen also proceed through the reaction easily to form coupling products in excellent yields. To get insight into the
mechanistic features, the authors have performed some control experiments, such as a radical clock, TEMPO and fluorescence quenching. Based on these experiments the authors proposed a feasible mechanism [Scheme 26].
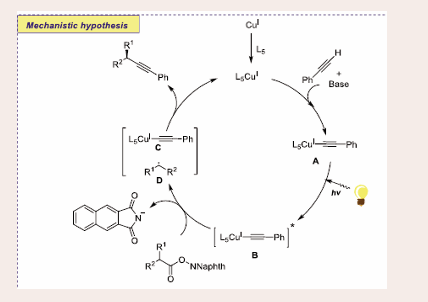
Scheme 26: Plausible mechanistic pathway for Cu-based Photocatalytic decarboxylative alkynylation reaction of alkynes and RAEs of carboxylic acids.
The reaction begins with the formation of CuI acetylene complex A through the reaction between the CuI catalyst, base and ligand. Afterward, this CuI acetylene complex A readily excited under photo-irradiation to afford excited complex B. Afterwards, SET between excited complex B and NHP-type ester releases CuII intermediate C. Next, radical decarboxylation of anionic radical of NHP ester generates alkyl radical D. Finally, alkyl radical D forms a chiral C(sp3)–C(sp) bond with CuII intermediate C to access the desired product and CuI catalyst is regenerated.
Recently, Yang and his colleagues demonstrated a dual copper photocatalyst and visible-light-mediated desulphurization strategy for the aliphatic C(sp3)–H alkylation incorporating Sonogashira coupling reaction [Scheme 27] [40].
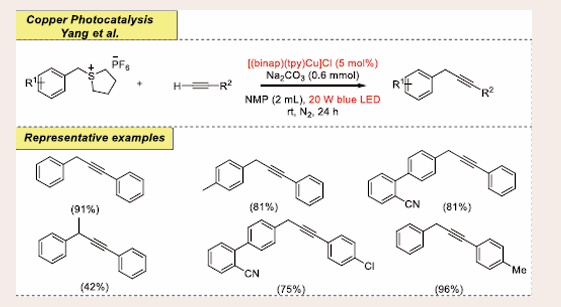
It was subjected to 1-benzyltetrahydrothiophenium hexafluorophosphate (0.4 mmol) as the source of sulfonium salt and phenylacetylene (0.02 mmol) under visible-light irradiation (20 W, 455 nm LED) with photocatalyst [(binap)(tpy)Cu]Cl (5 mol%) at room temperature for 24 hours in an NMP solvent and base (Na2CO3, 0.6 mmol). The desired coupling yields were obtained with excellent efficiency. In comparison to other bases like Cs2CO3, K2CO3, pyridine and Et3N; Na2CO3 is superior and furnishes the unsurpassed yield of the previously mentioned strategy. After the screening of various solvents, only NMP promotes the reaction competently. Also, scrutinized the scope of substrate concerning both benzylsulfonium salts and sulfonium salt under the optimized environments and detected good yields of the products. An extensive variety of substituents, such as Ph, BnO, Me, Br, F, ether and cyano moieties, on the benzylsulfonium salts is abided. This process is also operated well when sterically encumbered sulfonium salt was present with phenylacetylene which allows coupled product but-1yne-1,3-diyldibenzene in 42% yield. The coupling of benzylsulfonium salts with alkynes having various electron-rich and electron-deficient produces desired product in good to higher yields.
The possible transformation can ensue through the photocatalytic path as described in [Scheme 28].
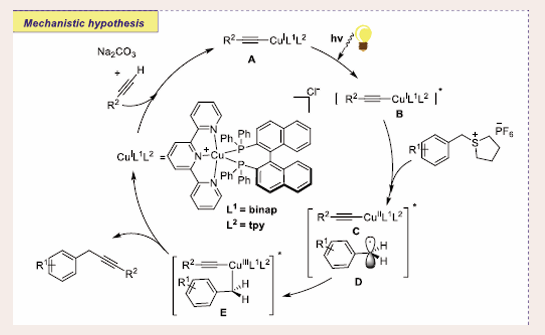
Scheme 28: Plausible mechanistic pathway of Cu-based photocatalytic Sonogashira couplings of sulfonium salt and phenylacetylene.
Firstly, the copper-complex A is generated through the reaction between [(binap)(tpy)Cu]Cl and alkynes. The resultant species A experiences photoexcitation and excited copper complex B is generated, which reacted with sulfonium salts to render complex C and the radical D. Simultaneously, intermolecular coupling of the CuII species C with D delivers the CuIII species E. At last, Sonogashira coupled products are being delivered by reductive elimination of species E and regenerating complex A, which is further anticipated in the catalytic cycle.
Catalytic C(SP2)-C(SP2) Bond Formation
In 2021, Hwang et al., unfolded a visible-light-promoted copper-photoredox-mediated aerobic oxidative annulation for Friedel–Crafts-type cyclization towards the synthesis of functionalized quinazolines through appropriate execution of coupling partners amidines and terminal alkynes [Scheme 29][41].
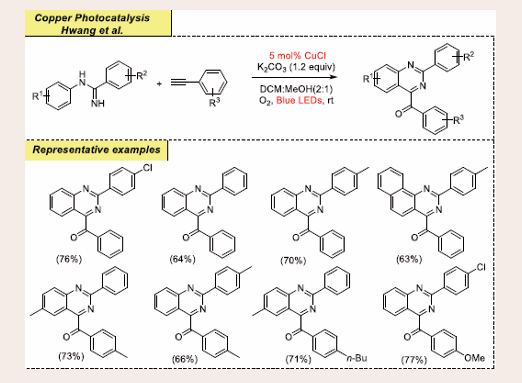
Scheme 29: Visible-light mediated copper-photoredox catalyzed Friedel–Crafts-type cyclization reaction.
They reported CuI-phenylacetylide catalyzed photo- oxidative Csp2–H annulation of amidines at room temperature, which is comparatively challenging to the conventional transition metal-catalyzed thermal annulation reactions. They performed the preliminary reaction using phenylacetylene and 4-chloro-N-phenylbenzimidamide as ideal substrates. Optimization screenings reveal the use of 5 mol% CuCl as catalyst, 1.2 equiv K2CO3 as base and molecular oxygen as an oxidant under in DCM: MeOH (2: 1) at room temperature under visible-light. In comparison to solvents like ACN, DCM, DCE, and THF; the combination of DCM and MeOH is superior and offers the unsurpassed yield for the aforesaid reaction strategy. An extensive variety of halogen substituents, electron-donating, withdrawing or neutral functionalities, such as ethyl, methyl, and even methoxy moieties, on various amidine and alkyne derivatives, is abided with moderate to good yields. This process is also operated well to produce anti-cancer compounds within 3 steps.
Based on a series of control studies, a catalytic sequence for this conversion is projected in [Scheme 30].
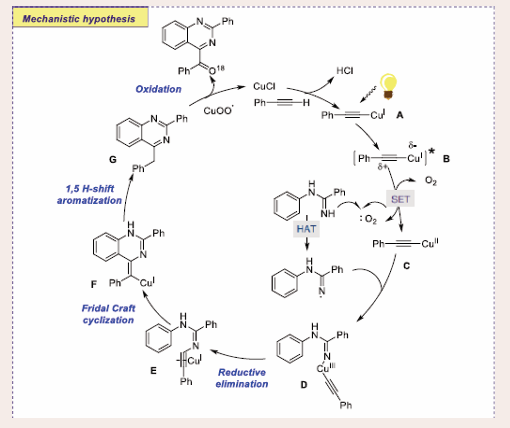
Scheme 30: Plausible mechanistic pathway for Cu-based Photocatalytic Friedel–Crafts-type cyclization of amidines and terminal alkynes.
The reaction starts with the generation of photoexcited triplet state CuI-phenylacetylide B complex from CuI-phenylacetylide A, which is followed by SET- mediated reduction by molecular oxygen to give CuII complex C and superoxide anion radical species. Subsequently, the copper- superoxo radical anion absorbs acid and reacts with complex C to generate CuIII-complex species D. On the other hand, the CuIII-complex D undergoes reductive elimination to generate ynamine intermediate E, which is followed by Friedel–Crafts- type cyclization to create intermediate F. Next, aromatization of cyclized intermediate produces compound G via 1,5-H-shift.
Finally, photo-oxidation of compound G derives the desired product by CuII superoxo. Furthermore, the present approach is straightforward, inexpensive, and does not create any hazardous byproducts that are detrimental to the ecosystem. It also does not need any costly, poisonous external photocatalysts or oxidants at room temperature. Additionally, according to calculations based on green chemistry metrics, the E-factor for the present green organic reaction is ∼ 1.9 times superior to that of the previously reported thermal technique.
Ackermann discovered a visible light-induced C–H arylation of azoles using a dual-catalytic system featuring an inexpensive, ligand-free copper(I) catalyst in combination with a photoredox catalyst. The organic photoredox catalyst 10-phenylphenothiazine (PTH) was identified as an effective, cost- efficient, and environmentally friendly alternative to the more commonly used, expensive IrIII-based complexes [Scheme 31] [42].
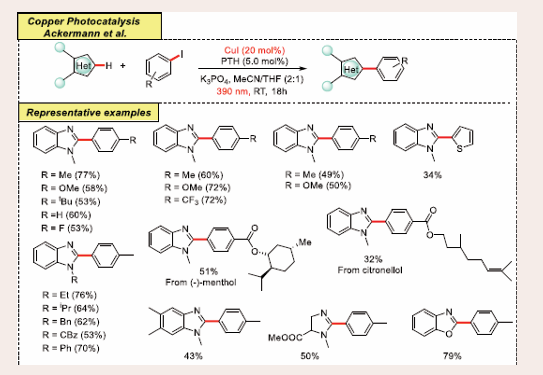
Scheme 31: Visible-light mediated copper-photoredox catalyzed C–H arylation of 1,3-azoles via copper/photoredox dual catalysis.
This method was successfully applied to the C–H arylation of various azole derivatives, including oxazoles, benzoxazoles, thiazoles, benzothiazoles, as well as more challenging substrates like imidazoles and benzimidazoles. The strategy's synthetic utility was demonstrated through the derivatization of complex molecules and the gram-scale synthesis of the natural product balsoxin. Mechanistic studies support a single electron transfer (SET) pathway, with an aryl radical as the key intermediate.
Using the optimized conditions, the photo-induced copper- catalyzed C–H arylation with PTH demonstrated versatility with a range of aryl iodides, both electron-donating and electron- withdrawing, achieving yields up to 77%. The mild conditions tolerated sensitive functional groups like nitrile, ester, and chloro- containing substrates, although with some proto dehalogenation. Additionally, complex molecules such as 2-iodothiophene and natural product-embedded aryl iodides were successfully transformed. The method also applied to various substituted benzimidazoles, imidazoles, and other heterocycles like benzoxazole, oxazoles, and thiazoles. Gram-scale synthesis of balsoxin further demonstrated the scalability, yielding 72%.
Based on literature precedents and mechanistic findings, a plausible mechanism for the photo-induced copper-catalyzed C–H arylation is proposed [Scheme 32].
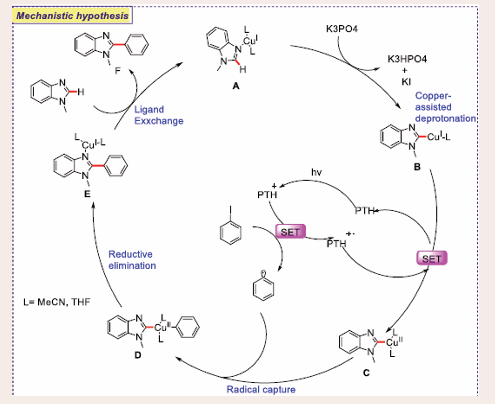
Scheme 32: Plausible mechanistic pathway of Photocatalytic C–H arylation of 1,3-azoles via copper/photoredox dual catalysis.
The reaction begins with the in-situ formation of copper complex A from copper(I) iodide, coordinated with the substrate and solvent molecules. Copper coordination at the Lewis-basic N(3) nitrogen of substrate increases the C–H acidity, facilitating copper-assisted C–H bond cleavage at the C(2) position to form copper complex B. Oxidative quenching of the excited PTH* species through a single electron transfer (SET) process generates an aryl radical, which is captured by copper complex C, formed by the oxidation of complex B, thereby regenerating the photoredox catalyst. The resulting CuIII complex D undergoes reductive elimination, forming the desired C–C bond. Ligand exchange with another benzimidazole molecule releases the product and regenerates the copper catalyst for further reactions.
Catalytic C(SP2)-C(SP) Bond Formation
In 2018, Hwang et al., magnificently achieved a simple and green approach for the preparation of 2- (arylethynyl /alkyl) pyridines via denitrogenative oxidative coupling with substrates terminal alkynes and hydrazinylpyridines using simple visible- light and copper complex as a photocatalytic system [Scheme 33] [43].
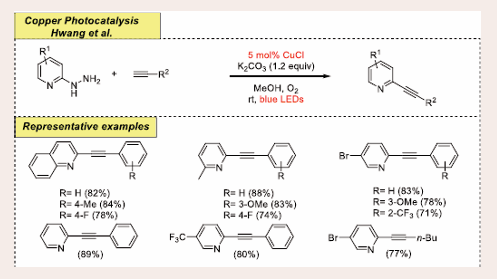
Scheme 33: Visible-light mediated copper-photoredox catalyzed denitrogenative oxidative reaction.
In this coupling reaction, 2-hydrazinylpyridine coupled with phenylacetylene in the existence of 5 mol% of CuCl catalyst, 1.2 equiv of K2CO3 base and molecular oxygen oxidant in methanol dispenses pyridines derivatives in excellent yield under blue LED irradiation for 10 hours. Changing copper with different metal catalysts, such as cobalt, nickel, iron, and zinc catalysts (viz, CoCl2, NiCl2, FeCl2, ZnCl2), gives rise to diminished titled product generation. In the substrate scope study, both coupling partners were successfully demonstrated under standard conditions. The study was carried out in the broad range of 2-hydrazinylquinoline bearing methyl, fluoro, methoxy at ortho, meta and para positions. A gram-scale reaction carried out under normal circumstances, significantly increases the synthetic usefulness of this coupling reaction. Green chemistry metrics demonstrate that this procedure is environmentally friendly and very cost-effective.
Based on control experiments and radical trapping experiments using TEMPO, the authors suggested possible mechanisms [Scheme 34].
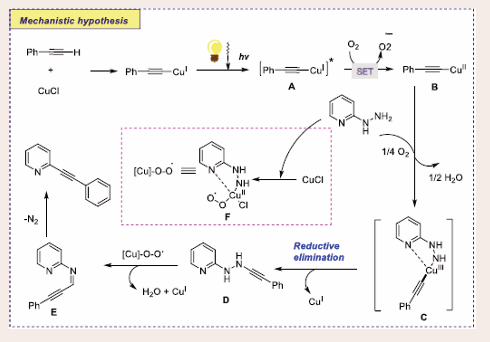
Scheme 34: Plausible mechanistic pathway for Cu-based Photocatalytic denitrogenative oxidative reaction of terminal alkynes and hydrazinylpyridines.
Initially, excited state of *CuI complex is generated through photo-irradiation on CuI phenylacetylide complex. CuII complex B can be afforded via single electron transfer reduction of on CuI phenylacetylide complex and superoxide radical anion. Later on, CuIII intermediate C species may be generated through nucleophilic addition of 2-hydrazinopyridine to CuII complex B. Next, reductive elimination separates the complex D with the generation of CuI species from CuIII intermediate C. 2-Hydrazinopyridine reacts with CuI and molecular oxygen to finish up the synthesis of the chelated bidentate CuII peroxo/- superoxo complex F, It abstracts the more acidic protons from the complex D to afford complex E. At last, the desired coupling product is obtained by nitrogen elimination of complex E, which leads to the combination of two radical species to dispense the final product.
CARBON-HETEROATOM BOND FORMATION
An energetically demanding substrate activation phase has prevented the widespread adoption of copper-catalyzed carbon- hetero bond formation by means of synthesizing medicinally relevant organic compounds (oxidative addition into an aryl halide). For the creation of C-X bonds, combining copper catalysis with photoredox catalysis has proven a promising strategy.
Catalytic Carbon−Nitrogen Bond Formation
In the midset of 2018, a straightforward strategy to access biologically fascinating alpha azido ketones via radical oxo- azidation of vinyl arenes under visible-light and the copper catalyst was introduced by the Reiser group [Scheme 35] [44].
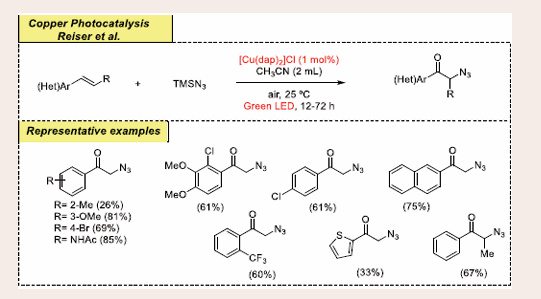
Scheme 35: Visible-light mediated copper-photoredox catalyzed oxo-azidation reaction.
The initial transformation has been achieved by using readily available styrene and TMSN3 under aerobic conditions. The optimized conditions for this reaction are as follows: 0.5 mmol vinyl arenes, 1.0 mmol TMSN3, 1 mol% of [Cu(dap)2]Cl as a catalyst in 2 mL methyl cyanide under 530 nm green LEDs irradiation. They found that the presence of copper catalyst and light irradiation is essential for getting the desired coupling products. Olefin containing both electron-efficient and electron- deficient substituents such as (-NH , -CN, NO , -COCH , -alkyl and -alkoxy) at the meta or para position of the benzene ring react quite smoothly and well, dispensing average to good yields. Remarkably, heteroaryl-substituents on olefin are tolerated nicely with excellent yield. There are several notable advantages to this reaction including its wide substrate scope, straightforward technique, inexpensive copper catalyst, step- economical process and oxidant-free approach.
To get insight into the mechanistic features, the authors performed several control experiments, including radical trapping experiments using TEMPO [Scheme 36].
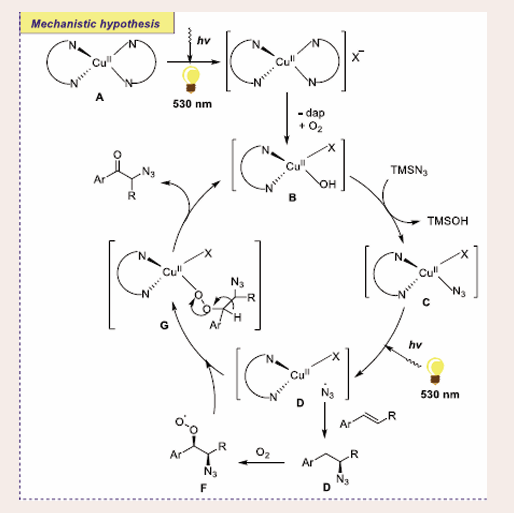
Scheme 36: Plausible mechanistic pathway for Cu-based Photocatalytic oxo-azidation reaction of vinyl arenes.
The copper catalyst, [Cu(dap)2]Cl A, first undergoes photoexcitation under green LEDs to yield an excited *CuI complex B, followed by homolytic dissociation of species C to yield CuI complex D and azide radical. Simultaneously, the addition of an azide radical to the olefin renders a stable radical E. Then radical E coordinates with molecular oxygen and affords radical F. Oxygen-centered radical can readily bind with radical E to release CuII complex G. Finally, the elimination of *CuI complex B from CuII complex G allows the final alpha azido ketone product.
In 2019, Zhang et al., disclosed a three-component applied route for intermolecular carboamination of alkenes, carbohalides, and amines [Scheme 37] [45].
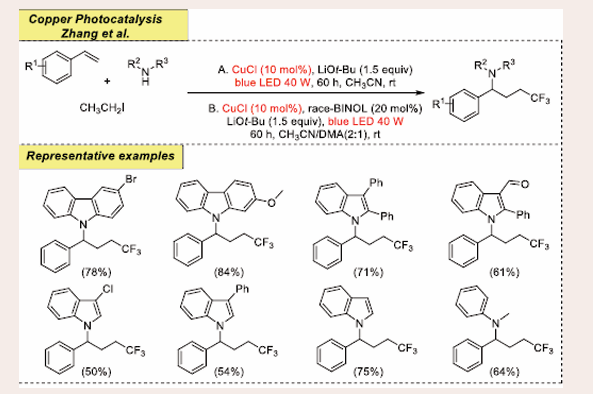
Scheme 37: Visible-light mediated copper-photoredox catalyzed intermolecular carboamination reaction.
The optimal reaction condition contains, 10 mol % CuCl as the catalyst, 1.5 equiv of LiOt-Bu as the base, 1.5 ml of CH3CN as solvent at room temperature along with radiation source blue LEDs (40 W), for 60 hours. Importantly, CuCl is utilized as both photo- and coupling catalyst for this transformation. Under the optimized conditions, a broad range of substrates achieve the desired coupling product. Electron-withdrawing and electron-rich styrenes are efficiently tolerated. Interestingly, 2-iodo-1,3-dimethylbenzene is termed to be an inefficient coupling moiety. The authors also anticipated that such a universal platform will be used for a wide range of functionalization of amines in medicinal chemistry due to the commercial availability of parent precursors as well as the relatively moderate conditions. Based on control studies such as radical-clock experiments and radical trapping experiments, the authors suggested a proposed mechanism. The mechanism involves CuI - *CuI - CuII - CuIII - CuI conversion of the copper catalyst via single electron transfer.
Encouraged by the aforementioned results, the same research group has also developed a straightforward tactic for the intermolecular Markovnikov hydroamination of olefin with amines as aminating agents under visible-light-mediated copper-photoredox catalysis [Scheme 38][46].
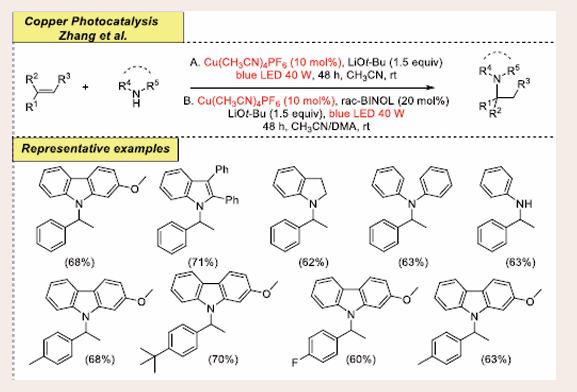
Scheme 38: Visible-light mediated copper-photoredox catalyzed Markovnikov hydroamination reaction.
They have accomplished the initial reaction using 9H-carbazole, 2-bromo- 2-methylpropane, and styrene as benchmark substrates. Optimization studies reveal that 10 mol % Cl(CH3CN)4PF6 catalyst, 1.5 equiv of LiOt-Bu a base, 2.0 ml of CH3CN solvent at room temperature along with radiation source blue LEDs (40 W), for 48 hours at room temperature furnishes the products. Since commercially available chemical substrates, operational simplicity and relatively moderate temperatures make this process more efficient than other methods. Even after an extended reaction time, the final yield is drastically reduced without blue LEDs irradiation. The technique has an acceptable scope in terms of the substrate. The electron-poor and electron- rich functionalities at the ortho and para positions of the phenyl ring generate compounds with high to average yields under optimal reaction conditions. Moreover, the amine substrates, including 9H-carbazole, indole derivatives, diphenylamine and aniline derivatives, produce the coupling products in modest to good yields.
Initially, in the basic medium, Cu-Nu complex B is generated through ligand exchange, which is reacted with an alkene to furnish the species C. Excited copper complex D is afforded through photo-irradiation of species C. Later on, excited copper complex D is readily converted into hypervalent copper complex F and affords benzyl radical E via SET and subsequent abstraction of a proton from methyl cyanide. Finally, radical E is trapped by the hypervalent copper complex F to furnish a hydroamination product [Scheme 39].
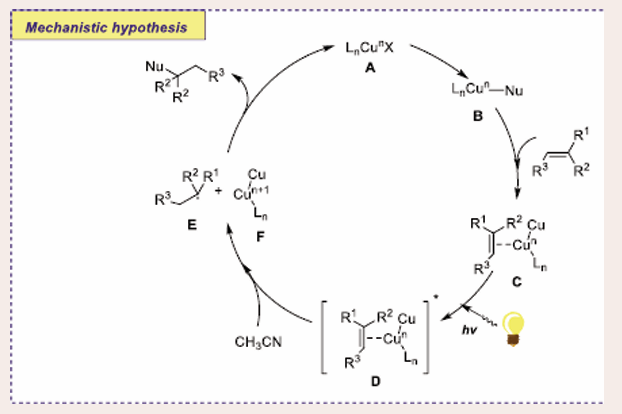
Scheme 39: Plausible mechanistic pathway for Cu-based Photocatalytic Markovnikov hydroamination reaction of olefine with amines.
According to Hwang et al., visible-light-promoted Cu- catalyzed hydrogen atom transfer (HAT) is a successful approach for synthesizing propargylamine derivatives from substituted amines and alkynes. [Scheme 40] [47].
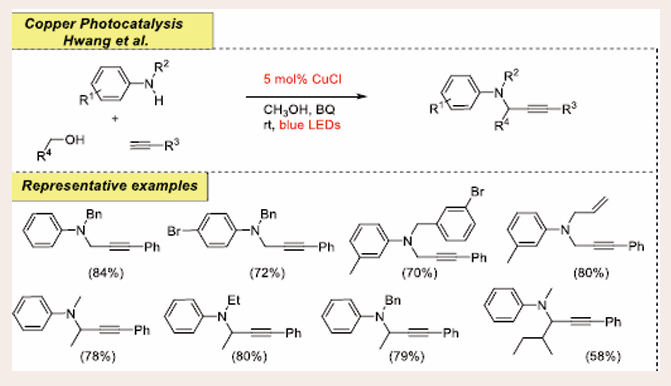
Scheme 40: Visible-light mediated copper-photoredox catalyzed hydrogen atom transfer reaction.
N-ethylaniline and phenylacetylene are used as preliminary substrates, which have been subjected to reaction at room temperature using methanol as the solvent, 5 mol% CuCl as the copper-catalyst, and benzoquinone (BQ) as the oxidant. Substrates with electron- withdrawing substituents are found to provide moderate to high yields. Simultaneously, electron-releasing and neutral group-bearing moieties yield the necessary products at high concentrations. However, the Sonogashira reaction is improved in this coupling process using halo-substituted N-benzyl/alkyl anilines. In addition, the tetrahydroquinoline substrate was discovered to be a good substrate for this process. Branched aliphatic alcohols, 2-ethoxy-ethanol, cycloalkyl carbinols, 2-phenyl ethanol, and tetrahydro-3-furanmethanol all responded efficiently in this procedure to get the product in high to moderate yields. However, methanol generated higher quantities of propargylamines than long-chain aliphatic alcohols, indicating that the α-oxy radical species is involved through the hydrogen atom transfer (HAT) mechanism.
Using several radical scavenger experiments, mechanistic investigations reveal that the reaction involved a radical process. As indicated in [Scheme 41].
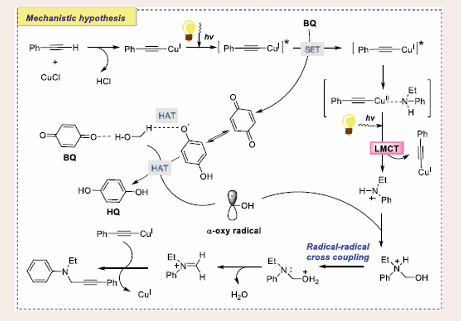
Scheme 41: Plausible mechanistic pathway for Cu-based Photocatalytic hydrogen atom transfer reaction of substituted amines and alkynes.
First, a triplet photoexcited CuI-phenylacetylide B complex is formed by in situ photo- irradiation and produces CuI–phenylacetylide A. Afterwards, the intermediate B may serve as an electron donor to BQ, resulting in CuII–phenylacetate C and a BQ radical anion E. It may weaken the α-C–H bond through the H-bonding interaction between BQ and OH group of CH3OH and HAT from C–H bond of CH3OH, which results in the creation of α-oxy radicals. Now, secondary arylamine then binds to the CuII site of intermediate C to form CuII–amine species D. An aminyl radical cation G and a stable CuI–phenylacetylide A are formed upon photoexcitation of the complex D. An iminium intermediate J is formed via an intramolecular proton transfer from water, via radical–radical cross-coupling. Ultimately, intermediate J and intermediate A are combined to produce the required product and regeneration of CuCl.
In 2023 Xei et al., discovered selective defluorinative functionalization of the trifluoromethyl group (–CF?) which offers an attractive route to fluorine-containing pharmaceuticals. Xie and co-workers reported a photoexcited copper-catalyzed strategy to activate the C–F bond of di- or trifluoromethylated arenes for divergent radical C–N coupling with carbazoles and aromatic amines. [Scheme 42][48].
![Scheme 42: Visible-light mediated copper-photoredox catalyzed C–N coupling with trifluoromethylated arenes. CuCl get back [Scheme 45].](https://www.jscimedcentral.com/public/assets/images/uploads/image-1738739314-1.PNG)
Scheme 42: Visible-light mediated copper-photoredox catalyzed C–N coupling with trifluoromethylated arenes. CuCl get back [Scheme 45].
Ligand variation allows product diversity, yielding α,α-difluoromethylamines from trifluoromethylated arenes and double defluorinative C–N coupling from difluoromethylated arenes. A carbazole-centered PNP ligand optimizes the copper-catalyzed C–N coupling to form imidoyl fluorides from aromatic amines. Additionally, a 1,2-difluoroalkylamination strategy for styrenes delivers γ,γ- difluoroalkylamines in synthetically useful yields. DFT studies reveal an inner-sphere electron transfer mechanism for selective activation of C(sp³)–F bonds.
With the optimized reaction conditions in hand, also explored the substrate scope. A variety of trifluoromethyl arenes afforded the desired C–N coupling products in moderate to good yields. Interestingly, arenes with electron-withdrawing groups at the para-position relative to the trifluoromethyl group hindered the reaction when the alkylphosphine ligand (nBu?P) was used, suggesting that electron-rich ligands disfavor the key coupling process. Conversely, electron-deficient ligands facilitated product formation. When the electron-poor phosphite ester ligand, P(OAr)? (Ar = 2,4-tBu?C?H?), replaced nBu?P, products were obtained in moderate yields. Benzotrifluorides yielded product only with 17% isolated yield. Additionally, α-(trifluoromethyl) styrene proved to be a suitable substrate, delivering product with good selectivity. Notably, difluoromethylated arenes were effective in Cu-catalyzed defluorinative C–N coupling, with double defluorinative processes leading to products with moderate yields.
According to plausible reaction mechanism by light irradiation, the catalyst [L1CuI] A is excited to [L1CuI]* B. This species can undergo either an outer-sphere single electron transfer (OSET) with the electron-deficient trifluoromethylated arene, forming [L1CuII]+ C and radical anion D, or an inner- sphere single electron transfer (ISET), yielding [L1CuII-F] E and the difluorobenzylic radical F. The radical anion from the OSET process loses a fluoride to generate the difluorobenzylic radical. When styrenes are used, the difluorobenzylic radical is readily converted to the more stable secondary benzyl radical G. Through ligand substitution, [L1CuII-F] transforms into [L1CuII- NHAr] H, which can interact with either radical to produce coupling products I or J. Although the difluorobenzylic radical or the benzyl radical is unlikely to undergo radical oxidative addition to generate a CuIII intermediate for subsequent reductive elimination, the possibility of forming coupling product cannot be ruled out. In particular, the NH in α,α-difluoromethylamines becomes more acidic due to the influence of the difluoromethyl group, facilitating the elimination of fluoride to produce imidoyl fluoride K as the final product [Scheme 43].
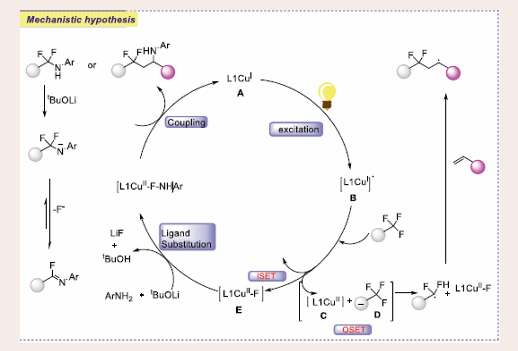
Scheme 43: Plausible mechanistic pathway for Cu-based Photocatalytic C–N coupling with trifluoromethylated arenes.
Catalytic Carbon−Sulfur Bond Formation
In a notable work, Anandhan and co-workers discovered an efficient one-pot approach for the construction of alkynyl sulfides, starting from 2-aminothiophenols and terminal alkynes. This novel protocol involves a one-pot visible-light-promoted copper- catalyzed aerobic oxidative for the construction of a novel C(sp)–S bond [Scheme 44 and 45][49].
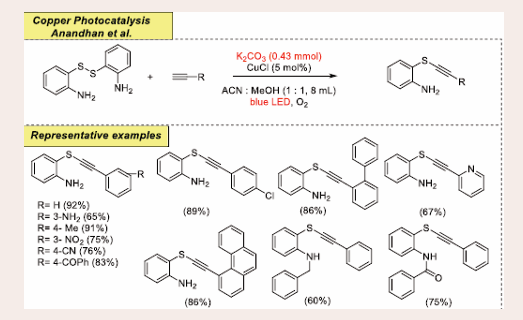
Scheme 44: Visible-light-mediated copper-photoredox catalyzed aerobic oxidative C(sp)–S coupling reaction.
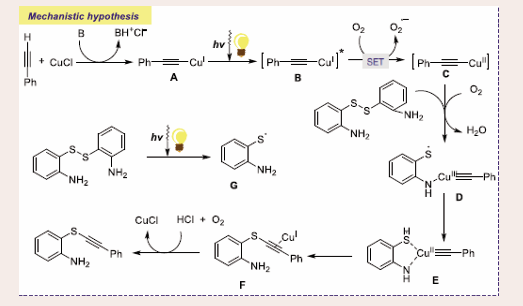
Scheme 45: Plausible mechanistic pathway for Cu-based photocatalytic aerobic oxidative C(sp)–S coupling of 2-aminothiophenols and terminal alkynes.
The preliminary reaction was performed using a dimer of 2-aminothiophenols and phenylacetylenes as model substrates. The optimized reaction conditions for C-S coupling were achieved using K2CO3 as base and CuCl (5 mol%) as a catalyst in an equivalent mixture of ACN (acetonitrile) : MeOH (1 : 1, 8 mL) under blue light. In this process, various functional groups on alkynes such as chloro, bromo, nitro, cyano, methyl, and methoxy are endurable well, the electronic and steric nature of the substituents do not show an obvious effect on the yield of the target product. In addition to naphthalene and anthracene, substituted alkynes are also found to be suitable substrates for this transformation. The current methodology featured several advantages such as simple starting materials, excellent chemoselectivity, oxidant-free, reduced synthetic steps, and high efficiency.
Mechanistically, the reaction begins with the photoexcitation of CuI-phenylacetylide complex A to achieve active CuI- phenylacetylide complex B, which furnishes CuII-phenylacetylide complex C and superoxide radical anion through SET with molecular oxygen. Consequently, photoexcitation of dimers of 2-aminothiophenols through visible-light deliver thiol radicals G. Later on, nucleophilic addition to CuII-phenylacetylide complex C of the amino group to access the CuII species D, which combines with the thiol radical to dispense CuII-aminothiophenol species E. Finally, species E coordinates with hydrochloric acid and molecular oxygen to release the desired C–S coupled product and catalyst
As a continuous study on the selective direct C– S bond formation using metallophotoredox catalysis, Reiser et al., reported copper−phenanthroline-based atom transfer radical addition (ATRA) reaction of alkenes and alkynes with p-toluenesulfonyl chloride under visible-light irradiation assembled chlorosulfonylated product [Scheme 46][50].
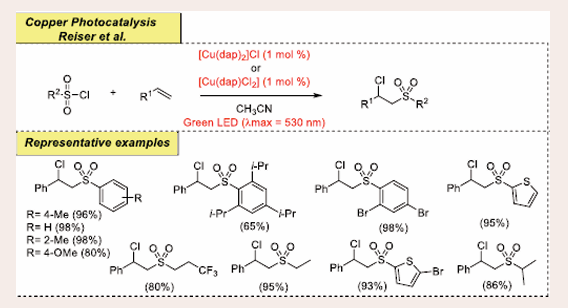
Scheme 46: Visible-light-mediated copper-photoredox catalyzed chlorosulfonylation reaction.
The study was initiated by examining the reaction of p-toluenesulfonyl chloride and styrene as the preliminary substrates. They used a catalytic system such as [Cu(dap)2]Cl as metal-photocatalyst under 530 nm irradiation. Interestingly, utilization of Na2CO3 (1 equiv) increases the desired product instead of K2CO3. In this protocol, sulfonyl chlorides having both electron-donating and electron-withdrawing groups participate a well in the reaction. Additionally, α- or β-alkyl substituted styrene also efficaciously undergoes this transformation and afforded the expected products in good yields. When Benzylic or allylic chlorides are exposed to styrene under reaction conditions, the desired products are obtained with satisfactory yields. Unfortunately, this current catalytic system is not fruitful for unactivated olefins. To demonstrate the generality and scalability of the strategy, a mmol scale reaction is also conducted. On the basis of the control experimental results, a plausible mechanism is proposed and depicted in [Scheme 47].
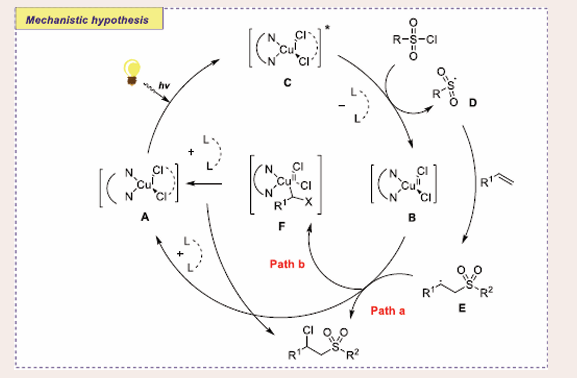
Scheme 47: Plausible mechanistic pathway for Cu-based photocatalytic chlorosulfonylation of alkenes.
The reaction is initiated with the decomposition of CuII−Cl to produce CuI intermediate A. Later, excited state of intermediate A, forms sulfonyl radicals D via the reduction of one-electron from sulfonyl chloride. Next, the radical D is added to an olefin to afford the radical intermediate E. Binding of intermediate E with intermediate B (persistent radical) transforms into F. Finally, the reductive elimination of intermediate F leads to the formation of the target product.
The heteropolycycles have the potential to be used as therapeutic probes and reactive intermediates. In 2021, Hwang and co-workers took advantage of the reactivity of the dual catalytic system consisting of CuI/ TMS-N3/visible-light to accelerate oxy-sulfonylation for the synthesis of new C-S bond containing β-keto sulfones as anti-analgesic agent [Scheme 48] [51].
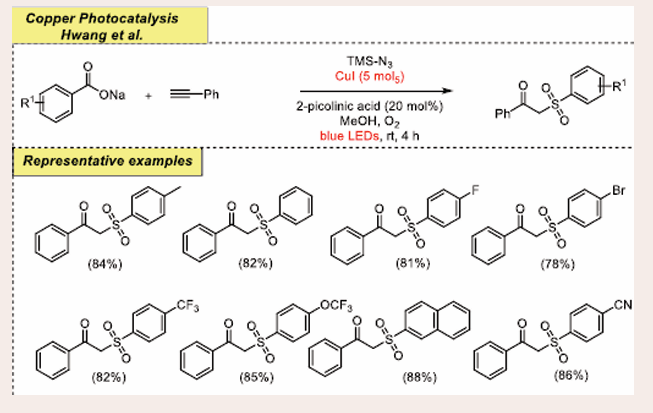
Scheme 48: Visible-light-mediated copper-photoredox catalyzed oxy-sulfonylation reaction.
Oxidative coupling reaction effectively furnishes the target product under standard conditions [CuI (5 mol %) and 2-picolinic acid (20 mol%) in a mixture of CH3CN:MeOH (1 : 1 v/v), 4 h]. This strategy unlocks a convenient and practical way for the synthesis of a new molecular framework containing substituted p-toluene sulfonate and well-decorated phenylacetylene in moderate to good yields. It is found that the substrates with electron-withdrawing groups (such as cyano, halo and nitro) on the sulfinate dispense the desired product in better yields. The initial components used in this approach are readily accessible and stable. Additionally, a variety of functional groups have been tolerated well by the presence of photoredox technique, avoiding the generation of side products. Based on this synthetic strategy, powerful physiologically active molecules including CES1, 11- HSD1 inhibitors, anti-analgesic drugs, and reactive intermediates were prepared.
According to the author's anticipated mechanism [Scheme 49],
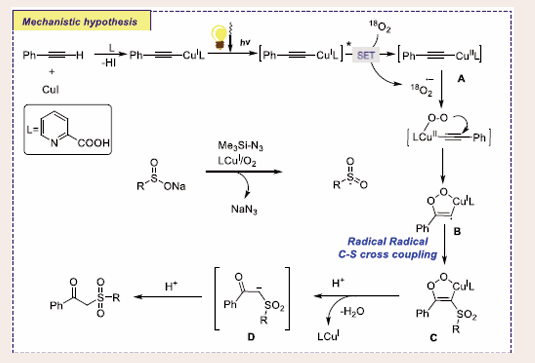
Scheme 49: Plausible mechanistic pathway for Cu-based photocatalytic oxy-sulfonylation of terminal alkynes.
the first stage of the reaction involves the formation of a triplet excited state CuI-phenylacetylide through photo-excitation of CuI-phenyl acetylide. Afterward, a single electron transfer (SET) process takes place, forming the intermediate A, followed by oxygen addition and radical-radical C-S coupling with sulfonyl radical form intermediate B. Lastly, the cleavage of O-O bond, followed by deprotonation from intermediate D furnishes the desired product β-keto sulfones. The new photochemical process is eco-friendly and extremely efficient, according to green chemistry metrics and Eco-scale assessment.
The Copper-catalyzed visible-light-induced ring-opening carbonylation of sulfonium salts introduces by Wu an innovative method that combines copper catalysis with visible light to achieve carbonylation reactions in 2023 [Scheme 50] [52].
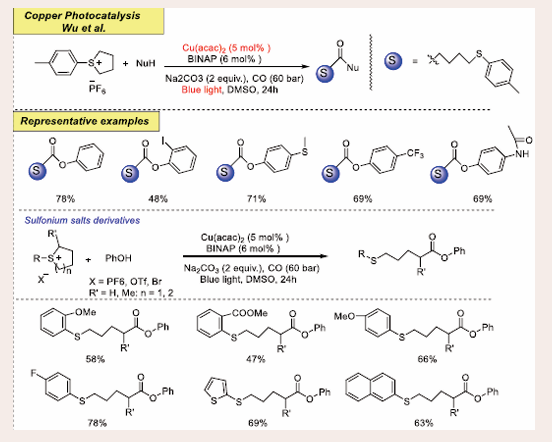
Scheme 50: Visible-light-mediated copper-photoredox catalyzed ring-opening carbonylation of sulfonium salts.
This process activates sulfonium salts, causing a ring-opening and subsequent carbonylation using carbon monoxide (CO) as the carbonyl source. The reaction proceeds under mild conditions, facilitated by visible light, and forms esters and amides efficiently. The use of copper as a catalyst makes the process cost-effective and environmentally sustainable. The reaction involves the generation of sulfonium-derived radicals through visible-light- induced photoredox catalysis, which then undergo carbonylation. The mild conditions and efficient radical generation make this approach attractive for green synthesis, especially in the pharmaceutical and agrochemical industries.
After optimizing the reaction conditions, various nucleophilic reagents were assessed, yielding products in 48–78%. Functional groups like amide, carbamoyl, and hydroxyl remained intact, demonstrating excellent chemoselectivity. Sulfonium salts bearing either electron-donating groups (such as o-OMe, p-OMe) or electron-withdrawing groups (such as o-CO?Et, p-F, p-Br) on the aryl ring afforded the corresponding products with yields ranging from 47% to 78%. Both thiophene- and naphthalene-modified sulfur salts underwent smooth reactions, yielding products and with 69% and 63% yields, respectively. Furthermore, both branched product and extended carbon chain product were successfully synthesized from their respective sulfur salts, involving the addition of a carbon center. Other types of sulfonium salts, including allyl, 2-phenylvinyl, benzyl, and n-butyl, were also investigated. However, only the allyl- linked sulfonium salt produced a product in low yield due to double bond migration, while other starting materials remained unreacted. Additionally, no desired product was detected when four- and seven-membered sulfonium salts were tested under the standard conditions.
Based on the results, a plausible radical-mediated mechanism for the copper-catalyzed ring-opening carbonylation of sulfonium salts is proposed, as illustrated in [Scheme 51].
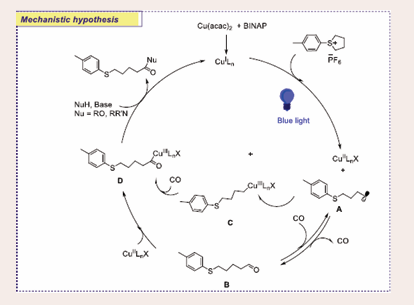
Scheme 51: Plausible mechanistic pathway for Cu-based photocatalytic ring-opening carbonylation of sulfonium salts.
The process begins with the formation of an active CuILn species generated in situ under the reaction conditions. Upon exposure to light, a single-electron transfer occurs between the CuILn complex and the sulfonium salt, producing CuIILnX species and an alkyl radical (A). The alkyl radical subsequently reacts with CO, forming an acyl intermediate (B), which can either be trapped by the CuIILnX species or rapidly recombine with the CuIILnX to yield intermediate C. In both pathways, insertion of CO into the C–Cu bond ultimately forms the acylcopper intermediate (D). Finally, nucleophilic attack on the acylcopper species produces the desired carbonylation product, regenerating the CuILn species to complete the catalytic cycle.
Catalytic Carbon−Oxygen Bond Formation
The radical-involved multicomponent difunctionalization of 1,3-dienes has emerged as an efficient strategy for synthesizing valuable allylic compounds in a one-pot operation by Chen in 2024 [Scheme 52] [53].
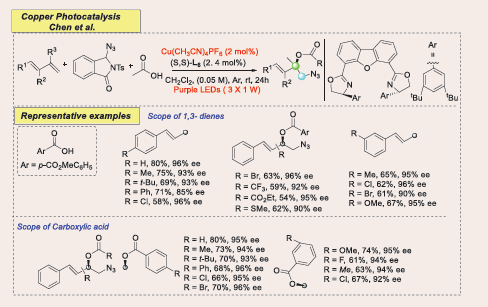
Scheme 52: Visible-light-mediated copper-photoredox catalyzed Radical 1,2-Azidooxygenation of 1,3-Dienes
However, challenges remain in expanding the scope of radicals and achieving enantiocontrol. This study presents a photoinduced copper-catalyzed, highly enantioselective three-component radical 1,2-azidooxygenation of 1,3-dienes, using readily available azido benziodazolone (Ts- ABZ) and carboxylic acids. The reaction exhibits broad substrate scope, high tolerance to functional groups, and excellent chemo-, regio-, and enantioselectivity, enabling the synthesis of azidated chiral allylic esters. Mechanistic investigations reveal that the chiral copper complex functions as a bifunctional catalyst, promoting both azidyl radical generation and enantioselective C–O cross-coupling. This one-step reaction effectively installs an azido group and oxygen-containing unit across C=C double bonds, greatly enhancing molecular complexity. While prior work in radical azidooxygenation provided high diastereoselectivity, catalytic asymmetric versions have been less explored, making this approach a novel contribution to the field.
An array of aryl-substituted 1,3-dienes bearing weakly electron-donating alkyl groups (e.g., Me, t-Bu) and aryl groups (e.g., Ph), as well as electron-withdrawing groups (e.g., Cl, Br, CF3, CO2Et, SMe) at the para-position of the phenyl ring, reacted smoothly, providing the corresponding 1,2-adducts in 54–75% yields with 85–96% enantiomeric excess (ee). Additionally, 1,3-dienes featuring various functional groups with diverse electronic and steric properties (e.g., Me, OMe, Cl, Br, F) at the meta- or ortho-position of the aromatic ring were also well tolerated. The desired cross-coupled products were obtained in 60–70% yields with 80–97% ee. Also, Benzoic acid and its derivatives, featuring electron-donating (e.g., Me, tBu, Ph) or electron-withdrawing (e.g., Cl, Br) groups at the para-position, reacted efficiently to yield products in 66–80% yields with excellent enantioselectivities (93–96% ee).
A plausible mechanism for the photoinduced bifunctional copper-catalyzed asymmetric three-component radical 1,2-azidooxygenation of 1,3-dienes is proposed. Initially, a photosensitive complex [L6/CuIO2CAr] B is formed, which, upon irradiation with purple LEDs, undergoes single electron transfer (SET) with Ts-ABZ, generating an azidyl radical and a CuII species C. The azidyl radical adds to the 1,3-diene, forming an allylic radical that is trapped by the CuII species, resulting in a π-allyl CuIII intermediate D. Reductive elimination yields the azidated chiral allylic ester, regenerating the CuI catalyst. This redox-neutral process proceeds under mild conditions with broad functional group tolerance, offering a sustainable, efficient method for synthesizing azidated chiral allylic esters. Future work aims to extend this strategy to other nitrogen-centered radical reactions [Scheme 53].
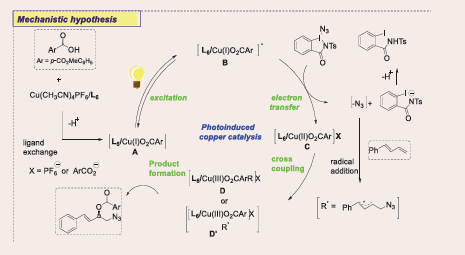
Scheme 53: Plausible mechanistic pathway for Cu-based photocatalytic radical 1,2-Azidooxygenation of 1,3-Dienes
Catalytic radical difunctionalization of 1,3-enynes has emerged as a promising method for synthesizing allenes and propargylic compounds discovered. While significant progress has been made in radical 1,4-difunctionalization, 1,2-difunctionalizations, particularly enantioselective ones, remain underexplored [Scheme 54] [54].
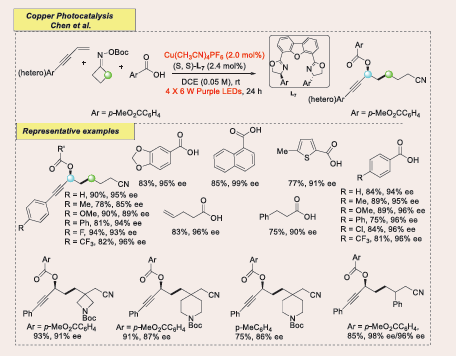
Scheme 54: Visible-light-mediated copper-photoredox catalyzed cross-coupling of 1,3-enynes with oxime esters and carboxylic acids
Herein, Chen report the first regio- and enantioselective radical three-component coupling of 1,3-enynes, oxime esters, and carboxylic acids via photoinduced copper catalysis. This redox-neutral protocol operates under mild conditions, showing broad functional group tolerance and 1,2-regioselectivity, yielding cyanoalkylated propargylic esters with excellent enantioselectivity (up to 99% ee).
Based on mechanistic studies and prior literature reports, a plausible mechanism proposed for this three-component radical cross-coupling reaction [Scheme 55].
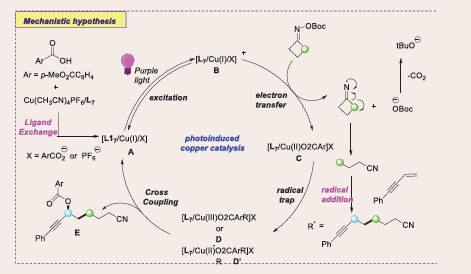
Scheme 55: Plausible mechanistic pathway for Cu-based photocatalytic cross-coupling of 1,3-enynes with oxime esters and carboxylic acids.
Initially, purple light irradiation activates a light-absorbing species, likely L17/CuI, A to its excited state B. This excited state undergoes a single electron transfer (SET) with the redox-active oxime ester, generating an iminyl radical and a CuII species III, with concurrent release of a carboxylate anion (BocO−). The iminyl radical undergoes β-C−C bond cleavage, producing a cyanoalkyl radical, which adds to the terminal carbon of the 1,3-enyne, forming a stabilized propargyl radical. The CuII complex III intercepts this propargyl radical, forming a CuIII intermediate D. Subsequent enantioselective reductive elimination yields the C–O cross-coupled product E. It's also possible that the Cu C complex D exists in equilibrium with a hybrid propargyl radical-type CuII complex D’, which may undergo an outer-sphere radical transfer or SET-oxidation. While these alternative pathways cannot be definitively ruled out and favour the CuI/II/III mediated catalytic cycle. Importantly, this chiral copper catalyst functions both as a photoredox catalyst and as the source of enantioselectivity in the C–O bond formation. The overall process is redox neutral, avoiding the need for external oxidants or reductants.
By Hu firstly regio- and stereoselective difluoroalkylthio- cyanation of alkynes with BrCF?R and KSCN has been reported under visible light-induced copper catalysis in 2024. The in situ formed copper complex not only generates CF?-alkyl radicals but also promotes C−SCN bond formation without additional photocatalysts or radical initiators. Internal alkynes are also transformed to CF?-derived tetrasubstituted olefins, with potential applications in agriculture and medicine [Scheme 56] [55].
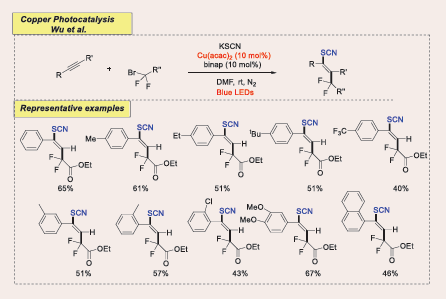
Scheme 56: Visible-light-mediated copper-photoredox catalyzed cross-coupling Regio- and Stereoselective Difluoroalkylthiocyanation of Alkynes
Electron-withdrawing groups showed good reactivity, yielding products with moderate to good yields and excellent regio- and stereoselectivity. Lower yield and stereoselectivity were likely due to scaling effects and prolonged reaction time. Hydroxymethyl was tolerated, yield with 38%. Halogen substituents were well tolerated and meta, ortho, and multisubstituted aryl rings produced with moderate yield. 1-Naphthyl and 6-quinolinyl ethynes formed with 46% and 43% yield while thiophene-derived acetylenes gave 48% and 44%. 1-Ethynylcyclohex-1-ene yielded the diene product, and bioactive L-menthol and L-borneol derivatives gave in moderate yields. Various bromodifluoroacetamides, including both acyclic and cyclic N,N-dialkyl amides, as well as N-monosubstituted amides, effectively generate CF2-alkyl radicals and facilitate the addition process demonstrating the reaction’s broad substrate scope with moderate yield.
According to proposed mechanism firstly, CuX is generated via disproportionation of CuX?. The copper complex A is formed in situ from CuX and binap, followed by ligand exchange with KSCN to yield photoactive [LCuISCN] B. Visible light irradiation generates the excited state III, which undergoes oxidative quenching with R Br to form the R radical and species D. The R radical reacts with alkynes to produce alkenyl radical E. Single- electron transfer from [LCuISCN] X to the alkenyl radical forms intermediate F, which undergoes reductive elimination to yield product G, regenerating [LCuIX] and completing the catalytic cycle [Scheme 57].
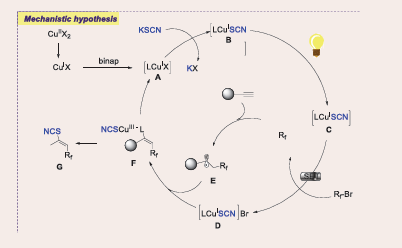
Scheme 57: Plausible mechanistic pathway for Cu-based photocatalytic Regio- and Stereoselective Difluoroalkylthiocyanation of Alkynes.
MISCELLANEOUS REACTIONS
In 2021, Bolm et al., improvised a visible-light-induced copper-photoredox-catalyzed chloro-sulfoximidation towards the synthesis of (E)-β-chlorovinyl sulfoximines through appropriate execution of coupling partners terminal aryl alkynes and sulfoximidoyl chlorides [Scheme 58] [56].
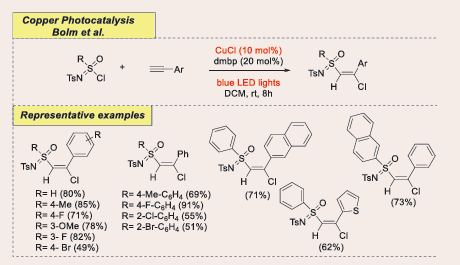
Scheme 58: Visible-light-mediated copper-photoredox catalyzed chloro-sulfoximidations reaction.
Initially, they performed stereo - and regioselectivity addition of N-tosyl protected sulfoximidoyl chloride to phenylacetylene as ideal substrates. Optimization screenings reveal that 10 mol% CuCl as a catalyst and 20 mol% dmbp (6,6-di-methyl-2,2-bipyridine) as a ligand in DCM at room temperature under visible-light for 8h furnishes product. In comparison to solvents like toluene, EtOH, 1,4-dioxane, CCl4, DCM, DMF, and THF; 1.2 ml DCM is superior and offers the unsurpassed yield for the aforesaid reaction strategy. An extensive variety of either halogen substituents, electron- donating, withdrawing or neutral functionalities, such as ethyl, methyl, and even methoxy moieties, on phenyl ring, is abided with moderate to good yields. Sometimes the ligand combination of dmbp and 1,10-phen provides a good amount of the desired addition reaction under standard conditions. Under an inert argon atmosphere, no transformation takes place.
Based on a series of control studies, a catalytic sequence for this conversion is projected in [Scheme 59].
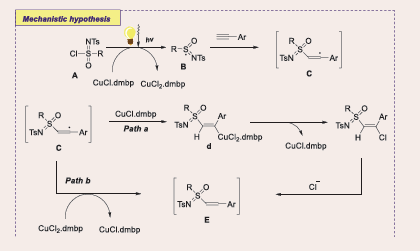
Scheme 59: Plausible mechanistic pathway for Cu-based photocatalytic chloro-sulfoximidations of terminal aryl alkynes
The reaction starts with the generation of sulfoximidoyl radical B through visible- light irradiation, which is readily added to alkyne to provide another radical complex C. According to the path, a, CuII-dmbp complex interacts with radical complex C to dispense trans-CuIII species D, which is followed by reductive coupling with chloride to offer E-isomer of the desired product. Consequently, path b also involves radical B. Oxidation of radical complex C takes place in the presence of CuII-dmbp complex to furnish cation E, which coordinates with chloride ion to access the desired addition product.
The arylethylamine pharmacophore is a key structure found in many biologically active natural products and pharmaceuticals, especially in compounds targeting the central nervous system. In this work, Ritter presented a photoinduced copper-catalyzed azidoarylation of alkenes using arylthianthrenium salts at a late stage, enabling the synthesis of highly functionalized acyclic (hetero)arylethylamine scaffolds that are otherwise challenging to obtain [Scheme 60] [57].
Scheme 60: Visible-light-mediated copper-photoredox catalyzed Late-Stage Azidoarylation of Alkenes via Arylthianthrenium Salts
The copper-catalyzed azidoarylation tolerates various complex arylthianthrenium salts, yielding functionalized acyclic (hetero) arylethylamines (40−71%) as a single isomer. It accommodates electron-rich, -neutral, or -poor substituents with para-, meta-, or ortho-substitution patterns. Azidoethyl groups can be introduced after converting arenes, phenols, and aryl chlorides to arylthianthrenium salts. Acrylonitrile yields unnatural (hetero) aromatic amino acids. Numerous functional groups, including halides, esters, and nitriles, are tolerated. A broad scope of alkenes, from Michael acceptors to unactivated ones, is compatible. Both electron-rich and electron-poor alkenes react well, enabling the synthesis of complex structures like adjacent quaternary centers and α-tertiary amines.
According to proposed mechanism shown in [Scheme 61].
Scheme 61: Plausible mechanistic pathway for Cu-based photocatalytic Late-Stage Azidoarylation of Alkenes via Arylthianthrenium Salts
The combination of rac-BINAP, Cu(MeCN)4BF4, and NaN3 in MeCN generates the photoactive copper catalyst B in situ. Upon excitation, catalyst B undergoes single-electron reduction of the arylthianthrenium salt, followed by mesolytic cleavage, producing an aryl radical and a copperII complex C or its cationic form with only one azide coordinated. The aryl radical adds to acrylonitrile, forming a homobenzyl radical, which then attacks the terminal nitrogen of the azido group in complex C via an outer-sphere pathway. This leads to the desired product while regenerating catalyst B. This C−N3 bond formation mechanism indicates potential for enantioselective azidoarylation using nonracemic BINAP-type ligands.
The combination of photoredox and copper catalysis has emerged as a novel approach in organic synthesis by Wui. In this study, Wui and co-workers represented an aminoalkylation of amino-dependent olefins with maleimides using a cooperative photo/copper catalytic system. This strategy enables the formation of a broad range of functionalized nitrogen- containing molecules, including oxazolidinones, 2-pyrrolidones, imidazolidinones, thiazolidinones, pyridines, and piperidines, without the need for an external photosensitizer or base [Scheme 62] [58].
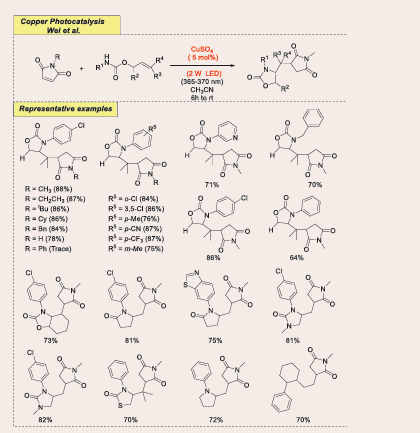
Scheme 62: Visible-light-mediated copper-photoredox catalyzed Aminoalkylation of Amino-Pendant Olefins
The method proceeds via a photoinduced CuI/CuII/CuIII catalytic cycle, involving nitrogen nucleophiles, intermolecular radical addition, and hydrogen atom transfer (HAT) processes. The proposed mechanism is supported by control experiments and theoretical studies, including radical scavenging, deuterium labeling, UV−vis absorption, and cyclic voltammetry (CV) analyses.
With the optimal conditions established, they explored the scope and limitations of the aminoalkylation. N-alkyl maleimides provided the desired products with electron releasing group with 84%–88% yields, while N-H maleimide 78% yield with Hydrogen derivatives. However, N-aryl maleimide failed to yield product with phenyl derivative, and acyclic electron-deficient olefins like diethyl maleate and styrene were unreactive. Then the scope of amino-dependent olefins studied on various amino-dependent olefins reacted smoothly with reagent, yielding products in 64%– 87%. Both electron-withdrawing and electron-donating groups on the arylamine moiety provided high yields of oxazolidinone derivatives. The N-heteroaryl substrate gave 71% yield, while the N-benzyl substrate yielded 70%, indicating that steric and electronic effects of N-substituents did not hinder the reaction. Additionally, 1,2-disubstituted and monosubstituted olefins were tolerated, producing moderate to good yields.
The proposed mechanism begins with the activation of substrate A by a CuII catalyst, leading to the formation of intermediate B. Upon light irradiation B undergoes an electronic excitation, forming reactive species C, which proceeds through nucleophilic addition to give intermediate D. Further transformations lead to the formation of E, which rearranges into cyclic intermediate F via a radical-mediated hydrogen atom transfer (HAT) mechanism. Finally, F converts to the product G, completing the catalytic cycle with the regeneration of the CuII catalyst in the presence of acetonitrile (CH?CN) [Scheme 63].
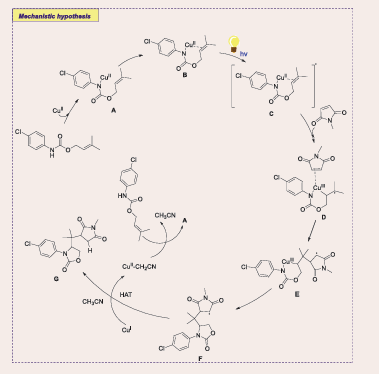
Scheme 63: Plausible mechanistic pathway for Cu-based photocatalytic Aminoalkylation of Amino-Pendant Olefins
A visible-light-promoted, CuI-catalyzed radical-triggered tandem cyclization of 1,7-dienes with cyclic sulfonium salts has been developed by Liu and co-workers. Using CuI-based photoredox catalysis under mild conditions, this method enables the synthesis of sulfur-containing polycyclic derivatives through sequential alkylation, 6-exo-trig, and 5-exo-trig cyclizations [Scheme 64] [59].
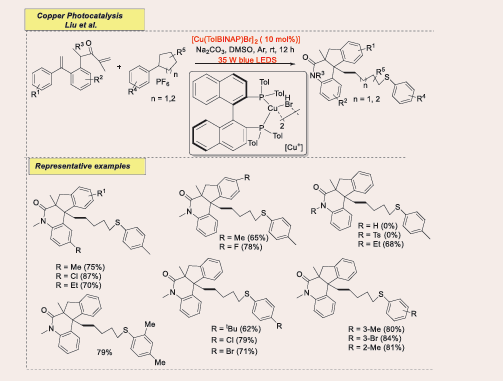
Scheme 64: Visible-light-mediated copper-photoredox catalyzed Alkylation/exo- Cyclization of 1,7-Dienes
The copper-catalyzed ring-opening of cyclic sulfonium salts generates alkyl radicals that selectively add to the C–C double bond adjacent to the aromatic ring, rather than to the electron-deficient vinyl group in 1,7-dienes. Additionally, oxidation of the sulfur-containing polycyclic products provides access to nitrogen-fused polycyclic derivatives with sulfoxide or sulfone groups, offering a versatile route to complex sulfur- and nitrogen-containing frameworks. A variety of N-methyl-N- (2-(1-phenylvinyl)-phenyl)-methacryl amides with electron- donating (Me, Et, OMe, tBu) and electron-withdrawing (F, Cl, Br, CN) substituents at different positions of the arylamine moiety smoothly underwent transformation under photoredox conditions, yielding sulfur-containing polycyclic products in 64%–91% yields. Halogen-substituted substrates converted efficiently, facilitating further modifications at the halogenated positions. Additionally, 1,7-dienes with substituents on the aromatic ring, including halogen or electron-donating groups (para-Me), yielded products in 65% and 78% yields, respectively. Methacrylamide, with an ethyl group on the nitrogen, gave the product in 68% yield, while N-H free and N-Ts substituted 1,7-dienes were unreactive.
Based on experimental results and literature reports, a plausible mechanism for the visible-light-induced Cu-catalyzed alkylation/cyclization of 1,7-dienes is proposed [Scheme 65].
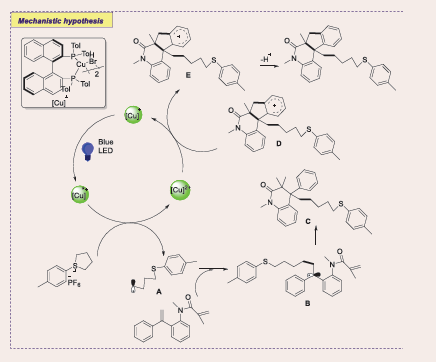
Scheme 65: Plausible mechanistic pathway for Cu-based photocatalytic Alkylation/exo- Cyclization of 1,7-Dienes
Upon 35 W blue LED irradiation, the photocatalyst [Cu]+ is excited to its [Cu]+* state. This excited species undergoes a single electron transfer (SET) with sulfonium salt, generating the CuII complex [Cu]2+ and an alkyl radical (A) via C(sp³)−S bond cleavage. The alkyl radical A selectively adds to the C=C bond connected to the aromatic ring in 1,7-diene, forming a stable benzyl radical (B). This selectivity, differing from previous studies, likely arises from the stability of the newly formed benzyl radical. The electron density and steric effects of the C=C bonds further influence this selectivity. A subsequent 6-exo-trig radical cyclization with the alkyne forms radical C, followed by a 5-exo- trig cyclization to produce intermediate D. A second SET occurs between [Cu]2+ and intermediate D, regenerating the ground- state [Cu]+ and forming the arenium ion E, which deprotonates to yield the final product.
CONCLUSION
This review is focused on the recent substantial developments which have been made in visible-light-promoted copper- catalyzed transformations. Over the past decades, the merging of metal catalysis and photoredox catalysis has surfaced as a diverse and powerful technique in the production of challenging molecular synthesis. Photoredox catalysis operated by visible light has recently emerged as a very powerful, safer, non-polluting, user-friendly alternative and plentiful renewable. Furthermore, copper complexes have been used as straightforward and cost-effective photoredox catalysts for a variety of organic transformations as a low-cost alternative to the complexes based on ruthenium or iridium. These photoinitiated Cu-catalyzed approaches have broad applicability, such as carbocyanation, trifluoromethylation, decarboxylative hydroalkylation, cyanation, alkynylation, hydrogen atom transfer reaction, and atom-transfer radical addition. Although Cu-based catalysts are currently experiencing significant success in photoredox catalysis. Several challenges are still needs to overcome. The oxidizing potential of Cu-catalysts involves transitions from *CuII to CuI or *CuI to Cu0, additional elongation of the excited state lifetime by advanced ligand design, and an increase in scale- up efficiency. In addition, ligand modification can efficiently tune copper-based photosensitizers by increasing coordinative stability in solution without sacrificing the favourable photo- physical properties of photosensitizers. Thus, it has been critical relevance to build novel copper-based compounds with enhanced redox and photophysical characteristics. In this regard, visible light-promoted Cu-catalysis still plays a crucial role in the growth of the novel stereoselective concept in many drugs, natural products, and other pharmaceutically relevant compounds. We anticipate that the information provided in this systematic review will inspire synthetic chemists to investigate the potential of visible-light-promoted copper-based transformations for future research.
ACKNOWLEDGMENTS
The authors are thankful to Veer Narmad South Gujarat University for providing all the necessary facilities.
REFERENCES
- Liu J, Xiao X, Lai Y, Zhang Z. Recent advances in transition metal- catalyzed heteroannulative difunctionalization of alkenes via C–H activation for the synthesis of heterocycles. Org Chem Front. 2022; 9: 2256-2279.
- Addison Ault. The Nobel Prize in Chemistry for 2001. Chem Educ. 2002; 79: 572.
- Charles P. Casey. 2005 Nobel Prize in Chemistry. Development of the Olefin Metathesis Method in Organic Synthesis. J Chem Educ. 2006; 83: 192.
- Suzuki A, Heck RF, Negishi E. Palladium-catalyzed cross-coupling reactions. TRENDS IN THE SCI. 2010; 399: 1811-1814.
- Yumeng xi, Hong Yi, Aiwen Lei. Synthetic applications of photoredox catalysis with visible light. Org Bio Che. 2013; 11: 2387-2403.
- Isaac Choi, Julia Struwe, Lutz Ackermann. C–H activation by immobilized heterogeneous photocatalysts. Photochem Photobiol Sci. 2021; 20: 1563-1572.
- Kirsten Zeitler Dr. Photoredox Catalysis with Visible Light. Angew Chem Int Ed. 2009; 48: 9785-9789.
- Jagan MR, Narayanam, Corey RJ. Stephenson. Visible light photoredox catalysis: applications in organic synthesis. Chem Soc Rev. 2011; 40: 102-113.
- David A Nicewicz, David WC MacMilla. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Sci. 2008; 322: 77-80.
- Jun Xuan, Prof. Dr. Wen-Jing Xiao. Visible-Light Photoredox Catalysis. Chem Int Ed. 2012; 51: 6828-6838.
- Harunobu Mitsunuma, Shun Tanabe, Hiromu Fuse, Kei Ohkubo, Motomu Kanai. Catalytic asymmetric allylation of aldehydes with alkenes through allylic C(sp3)–H functionalization mediated by organophotoredox and chiral chromium hybrid catalysis. Chem Sci. 2019; 10: 3459-3465
- Takashi Koike. Frontiers in Radical Fluoromethylation by Visible- Light Organic Photocatalysis. Asian J Org Chem. 2020; 9: 529-537.
- Chun-Jae Yoo, Daniel Rackl, Wenbin Liu, Caroline B Hoyt, Brian Pimentel, Ryan P Lively, et al. An Immobilized-Dirhodium Hollow- Fiber Flow Reactor for Scalable and Sustainable C-H Functionalization in Continuous Flow. Angew Chem Int Ed Engl. 2018; 57: 10923- 10927.
- Bryony M Hockin, Chenfei Li, Neil Robertson, Eli Zysman-Colman. Photoredox catalysts based on earth-abundant metal complexes. Catal Sci Technol. 2019; 9: 889-915.
- Xiaoqiang Huang, Eric Meggers. Asymmetric Photocatalysis with Bis- cyclometalated Rhodium Complexes Acc. Chem Res. 2019; 52: 833-847.
- Harshita Shet, Udaysinh Parmar, Shatrughn Bhilarea, Anant R. Kapdi. A comprehensive review of caged phosphines: synthesis, catalytic applications, and future perspectives. Org Chem Front. 2021; 8: 1599-1656.
- Zhengli Duan, Wu Li, Aiwen Lei. Nickel-Catalyzed Reductive Cross- Coupling of Aryl Bromides with Alkyl Bromides: Et3N as the Terminal Reductant. Org Lett. 2016; 18: 4012-4015.
- Dipannita Kalyani, Kate B. McMurtrey, Sharon R. Neufeldt, Melanie S. Sanford. Room-Temperature C–H Arylation: Merger of Pd-Catalyzed C–H Functionalization and Visible-Light Photocatalysis. J Am Chem Soc. 2011; 133: 18566-18569.
- Wissam Iali, Pierre-Henri Lanoe, Stéphane Torelli, Damien Jouvenot, Frédérique Loiseau, Colette Lebrun, et al. A Ruthenium(II)–Copper(II) Dyad for the Photocatalytic Oxygenation of Organic Substrates Mediated by Dioxygen Activation. Angew Chem. 2015; 127: 8535-8539.
- Allison G. CondieJosé, González-GómezCorey C, Stephenson RJ. Visible-Light Photoredox Catalysis: Aza-Henry Reactions via C−H Functionalization. J Am Chem Soc. 2010; 132: 1464-1465.
- Kelvin Pak Shing, CheungSumon Sarkar, Vladimir Gevorgyan. Visible Light-Induced Transition Metal Catalysis. Chem Rev. 2022; 122: 1543-1625.
- Aurélie Lavie-Cambot, Martine Cantuel, Yoann Leydet, Gediminas Jonusauskas, Dario M. Bassani, Nathan D. McClenaghan. Improving the photophysical properties of copper(I) bis(phenanthroline) complexes. Coord Chem Rev. 2008; 252: 2572-2584.
- Jérôme Beaudelot, Samuel Oger, Stefano Peruško, Tuan-Anh Phan, Titouan Teunens, Cécile Moucheron, et al. Photoactive Copper Complexes: Properties and Applications. Chem Rev. 2022; 122: 16365-16609.
- Sebastian Engla, Oliver Reiser. Copper-photocatalyzed ATRA reactions: concepts, applications, and opportunities. Chem Soc Rev. 2022; 51: 5287-5299.
- Jian He, Caiyou Chen, Gregory C. Fu, Jonas C. Peters. Visible- Light-Induced, Copper-Catalyzed Three-Component Coupling of Alkyl Halides, Olefins, and Trifluoromethylthiolate To Generate Trifluoromethyl Thioethers. ACS Catal. 2018; 8: 11741-11748.
- Xiao-Ye Yu, Quan-Qing Zhao, Jun Chen, Jia-Rong Chen, Wen-Jing Xiao. Copper-catalyzed radical cross-coupling of redox-active oxime esters, styrenes, and boronic acids. Angew Chem. 2018; 130: 15731- 15735.
- Jun Chen, Bin-Qing He, Peng-Zi Wang, Xiao-Ye Yu, Quan-Qing Zhao, Jia-Rong Chen, et al. Photoinduced, Copper-Catalyzed Radical Cross- Coupling of Cycloketone Oxime Esters, Alkenes, and Terminal Alkynes. Org Lett. 2019; 21: 4359-4364.
- Shi D, Guo X, Lai T, Zheng K, Wu Q, Sun C, et al. Rational Catalyst Design for N2 Reduction under Ambient Conditions: Strategies toward Enhanced Conversion Efficiency. J Inorganic Chem Commun. 2019; 105: 9-12.
- Bowen Han, Yanjun Li, Ying Yu, Lei Gong. Photocatalytic enantioselective α-aminoalkylation of acyclic imine derivatives by a chiral copper catalyst. Nat Commun. 2019; 10: 3804.
- Antoine Caron, Émilie Morin, Shawn K. Collins. Bifunctional Copper- Based Photocatalyst for Reductive Pinacol-Type Couplings. ACS Catal. 2019; 9: 9458-9464.
- Hang T Dang, Graham C Haug, Vu T Nguyen, Ngan TH Vuong, Viet D Nguyen, Hadi D Arman, et al. Acridine Photocatalysis: Insights into the Mechanism and Development of a Dual-Catalytic Direct Decarboxylative Conjugate Addition. ACS Catal. 2020; 10: 11448-11457.
- Nagarajan Ramkumar, Larisa Baumane, Dzintars Zacs, Janis Veliks, Angew. Merging Copper(I) Photoredox Catalysis and Iodine(III) Chemistry for the Oxy-monofluoromethylation of Alkenes. Chem Int Ed Engl. 2023; 135: e202219027.
- Sven Trienes, ab Jiawei Xua, Lutz Ackermann. Photoinduced C–H arylation of 1,3-azoles via copper/photoredox dual catalysis. Chem Sci. 2024; 15: 7293-7299.
- Marco M Mastandrea, Santiago Cañellas, Xisco Caldentey, Miquel A Pericàs. Decarboxylative Hydroalkylation of Alkynes via Dual Copper-Photoredox Catalysis. ACS Catal. 2020; 10: 6402-6408.
- Yue Jia, Zhihan Zhang, Guo-Ming Yu, Xuan Jiang, Liang-Qiu Lu, Wen- Jing Xiao. Visible Light Induced Copper-Catalyzed Enantioselective Deaminative Arylation of Amino Acid Derivatives Assisted by Phenol. Angew Chem Int Ed Engl. 2023; 135: e202312102.
- Yajing Zhang, Youwen Sun, Bin Chen, Meichen Xu, Chen Li, Dayong Zhang, et al. Copper-Catalyzed Photoinduced Enantioselective Dual Carbofunctionalization of Alkenes. Org Lett. 2020; 22: 1490-1494.
- Binlin Zhao, Yixiao Wu, bc Yu Yuanc, Zhuangzhi Shi. Copper-catalysed Csp3–Csp cross-couplings between cyclobutanone oxime esters and terminal alkynes induced by visible light. Chem Commun. 2020; 56: 4676-4679.
- Yu Mao, Wenxuan Zhao, Shuo Lu, a Lei Yu, Yi Wang, Yong Liang, et al. Copper-catalysed photoinduced decarboxylative alkynylation: a combined experimental and computational study. Chem Sci. 2020; 11: 4939-4947.
- Hai-Dong Xia, Zhong-Liang Li, Qiang-Shuai Gu, Xiao-Yang Dong, Jia-Heng Fang, Xuan-Yi Du, et al. Photoinduced Copper-Catalyzed Asymmetric Decarboxylative Alkynylation with Terminal Alkynes. Angew Chem Int Ed. 2020; 59: 16926-16932.
- Xuan Li, Min Jiang, Xiaolong Zhu, Xiuyan Song, Qirong Deng, Jian Lv, et al. A desulphurization strategy for Sonogashira couplings by visible light/copper catalysis. Org Chem Front. 2022; 9: 386-393.
- Vaibhav Pramod Charpe, Ayyakkannu Ragupathi, Arunachalam Sagadevan, Kuo Chu Hwang. Photoredox synthesis of functionalized quinazolines via copper-catalyzed aerobic oxidative Csp2–H annulation of amidines with terminal alkynes. Green Chem. 2021; 23: 5024-5030.
- Sven Trienes, Jiawei Xu, Lutz Ackermann. Photoinduced C–H arylation of 1,3-azoles via copper/photoredox dual catalysis. Chem Sci. 2024; 15: 7293-7299.
- Vaibhav Pramod Charpe, Aniket A. Hande, Arunachalam Sagadevan, Kuo Chu Hwang. Visible-light induced copper(i)-catalysed denitrogenative oxidative coupling of hydrazinylpyridines with terminal alkynes. Green Chem. 2018; 20: 4859-4864.
- Asik Hossain, Adiyala Vidyasagar, Christian Eichinger, Christian Lankes, Jenny Phan, Julia Rehbein, et al. Visible-Light-Accelerated Copper(II)-Catalyzed Regio- and Chemoselective Oxo-Azidation of Vinyl Arenes. Angew Chem Int Ed. 2018; 57: 8288-8292.
- Yang Xiong, Xiaodong Ma, Guozhu Zhang. Copper-Catalyzed Intermolecular Carboamination of Alkenes Induced by Visible Light. Org Lett. 2019; 21: 1699-1703.
- Yang Xiong, Guozhu Zhang. Visible-Light-Induced Copper-Catalyzed Intermolecular Markovnikov Hydroamination of Alkenes. Org Lett. 2019; 21: 7873-7877.
- Arunachalam Sagadevan, Kishore Kumar Pampana V, Kuo Chu Hwang. Copper Photoredox Catalyzed A3’Coupling of Arylamines, Terminal Alkynes, and Alcohols through a Hydrogen Atom Transfer Process. Angew Chem Int Ed. 2019; 58: 3838-3842.
- Jun Huang, Qi Gao, Tao Zhong, Shuai Chen, Wei Lin, Jie Han, et al. Photoinduced copper-catalyzed C–N coupling with trifluoromethylated arenes. Nat Commun. 2023; 14: 8292.
- Mandapati Bhargava Reddy, Ramasamy Anandhan. Visible light initiated amino group ortho-directed copper(i)-catalysed aerobic oxidative C(sp)–S coupling reaction: synthesis of substituted 2-phenylbenzothiazoles via thia-Wolff rearrangement. Chem Commun. 2020; 56: 3781-3784.
- Asik Hossain, Sebastian Engl, Eugen Lutsker, Oliver Reiser. Visible- Light-Mediated Regioselective Chlorosulfonylation of Alkenes and Alkynes: Introducing the Cu(II) Complex [Cu(dap)Cl2] to Photochemical ATRA Reactions. ACS Catal. 2019; 9: 1103-1109.
- Kishore Kumar Pampana V, Vaibhav Pramod Charpe, Arunachalam Sagadevan, Deb Kumar Das, Chun-Cheng Lin, Jih Ru Hwu, et al. Oxy- sulfonylation of terminal alkynes via C–S coupling enabled by copper photoredox catalysis. Green Chem. 2021; 23: 3569-3574.
- Zhang Y, Bao ZP, Kuai CS, Wu XF. Recent Progress in Synthesis, Mechanism and Applications of Zinc-Based Metal-Organic Frameworks for Fluorescent Sensing. J Catalysis. 2023; 426: 1-5.
- Guo-Qing Li, Zi-Qing Li, Min Jiang, Zhihan Zhang, Yu Qian, Wen-Jing Xiao, Jia-Rong Chen. Photoinduced Copper-Catalyzed Asymmetric Three-Component Radical 1,2-Azidooxygenation of 1,3-Dienes. Angew Chem Int Ed. 2024; 63: e202405560.
- Guo-Qing Li, a Fan-Rong Meng, Wen-Jing Xiao, Jia-Rong Chen. Photoinduced copper-catalyzed asymmetric radical three-component cross-coupling of 1,3-enynes with oxime esters and carboxylic acids. Org Chem Front. 2023; 10: 2773-2781.
- Hu X, Wang Y, Xu S, Wu J, Wu F. Hydrogenation of Sugar Enol- Ethers Using Pd/Et3SiH Reagent System: A Route to Deoxy Sugars/ Glycosides. The J Org Chem. 2024.
- Peng Shi, Yongliang Tu, Duo Zhang, Chenyang Wang, Khai-Nghi Truong, Kari Rissanen, et al. Regio- and Stereoselective Chloro Sulfoximidations of Terminal Aryl Alkynes Enabled by Copper Catalysis and Visible Light. ASC. 2021; 363: 2552-2556.
- Yuan Cai, Sagnik Chatterjee, Tobias Ritter. Photoinduced Copper- Catalyzed Late-Stage Azidoarylation of Alkenes via Arylthianthrenium Salts. J Am Chem Soc. 2023; 145: 13542-13548.
- Can-Can Zhang, Hong-Li Wu, Xuan-Chi Yu, Ling-Tao Wang, Yu Zhou, Yong-Bin Sun, et al. Photoinduced Copper-Catalyzed Aminoalkylation of Amino-Pendant Olefins. Org Lett. 2023; 25: 5862-5868.
- Xin-Qian Liu, Jian-Hong Fan, Ke-Wen Tang, Long-Jin Zhong, Yu Liu. Visible-Light-Induced Copper-Catalyzed Alkylation/exo- Cyclization of 1,7-Dienes with Sulfonium Salts to Access Sulfur- Containing Polycyclic Compounds. ASC. 2024; 366.









































































































































{kind=link}