Targeted Inhibitor Design: Lessons from Small Molecule Drug Design, Directed Evolution, and Vaccine Research
- 1. Department of Chemical and Biological Engineering, University at Buffalo, USA
ABSTRACT
Inhibitors of protein-protein interactions are useful for elucidating novel biology and for manipulating biological processes for therapeutic effects. To this end, both small molecule and protein inhibitors are commonly used. The discovery of novel binders is most often accomplished by screening a diversity oriented library. However, available biochemical and structural information on the target molecule is often neglected during the screen. Incorporating molecular details of the binding surface can improve the efficiency of discovering molecules with useful biological properties. We discuss how structural and other molecular data may be leveraged to bias high throughput screen and optimize the efficiency of identifying high affinity, functional binders. Examples from small molecule drug design, protein engineering and vaccine research are used to illustrate the point.
CITATION
Park S, Mann J, Li N (2013) Targeted Inhibitor Design: Lessons from Small Molecule Drug Design, Directed Evolution, and Vaccine Research. Chem Eng Process Tech 1: 1004.
INTRODUCTION
Protein-protein interactions (PPI) are essential for most biological processes, including signaling, transcription, metabolism, and proliferation. As such, an efficient method of targeting specific protein-protein interactions has numerous potential applications in basic science and medicine. For example, PPI inhibitors may be used to investigate the physiological roles of individual proteins and the biological pathways that depend on them. Targeting PPI is also therapeutically relevant since it may be used to reverse the disease phenotypes resulting from errant protein-protein interactions. Even when a disease causing mutation does not alter the biophysical attributes of a protein directly it may still result in changes in protein-protein interaction. For example, cancer causing mutations can occur in the promoter region and change the gene expression level without affecting the details of molecular interaction involving the gene product [1,2]. A PPI inhibitor can therefore have therapeutic efficacy against various of disease-inducing mutations, and an ability to design inhibitors to target specific protein-protein interactions has far reaching implications for a broad range of biological and medical problems.
While the benefits of designing epitope specific inhibitors are clear, there are significant challenges associated with the design of such molecules. In addition to the general pharmacological constraints for potential drugs, such as bioavailability and toxicity, there are limitations that are specific to each class of molecules. The difficulties of designing small molecule inhibitors of PPI have been described before [3,4]. Effective small molecule inhibitors are rare because protein-protein interfaces tend to include large surface areas, which are difficult to disrupt using small compounds. Also, the surfaces involved in transient protein-protein associations are typically solvent exposed and relatively flat--a combination of physical and chemical properties that does not easily render to high affinity recognition. Two strategies are often used for small molecule inhibitor design. A top down discovery strategy relies on high throughput screens (HTS) of large diversity oriented compound libraries [5]. Because potential drugs, and drug leads, are “discovered” from HTS, the success rate of such screens increases with the size of the library. The overhead of maintaining a large chemical library and the time it takes to assay their activity limits the maximum size of a library that can be practically screened to around 106 compounds.
The alternative, bottom up approaches include computational design [6], rational chemical synthesis, and fragment based ligand assembly [7]. These techniques seek to incorporate existing biochemical or structural data of the target molecule in order to maximize the probability of engineering molecules with useful functional properties. This is accomplished by explicitly targeting the surfaces known to be functionally important. As a result, the drugs are “engineered” during a bottom up approach. Because the molecules that bind at functionally relevant sites are also likely to have functional significance, judicious application of the strategy can lead to discovery of novel drugs starting from a library of much smaller diversity than typical for HTS.
Compared to the screening of small molecule libraries, which often requires robotics and automated analysis systems, protein inhibitors can be engineered without the use of an expensive experimental setup and can be readily applied to both research and therapeutic applications. Because they are straightforward to design and characterize, protein inhibitors may be used to investigate signaling pathways, validate drug targets, or generate a drug lead. The structural and functional properties of a protein can be changed by introducing mutations in DNA, which is an important distinction from small organic compounds. Protein binders have several advantages over small molecule inhibitors [8,9]. Because protein inhibitors bind their targets using a larger contact surface, they may be more effective in disrupting PPI. The larger interface of a PPI also allows fine tuning of the specificity of interaction and increases selectivity for their targets. Protein inhibitors may be engineered either rationally or through directed evolution [10]. There are several protein engineering platforms currently available for use [11,12].
Similarly to small molecule drug discovery, both top down and bottom up approaches are used for the engineering of protein inhibitors. For example, they may be computationally designed [13], or otherwise rationally engineered based on biochemical and structural data. Progress in computational protein design enables its use as an important complement to, if not a replacement of, an experimental screen. The traditional use of a diversity oriented library for the selection of protein binders is similar to that of small molecule HTS in that the binding affinity is engineered without explicit incorporation of the binding site information. That is, selection from a random library is performed without biasing the interaction toward a specific surface patch (Figure 1).
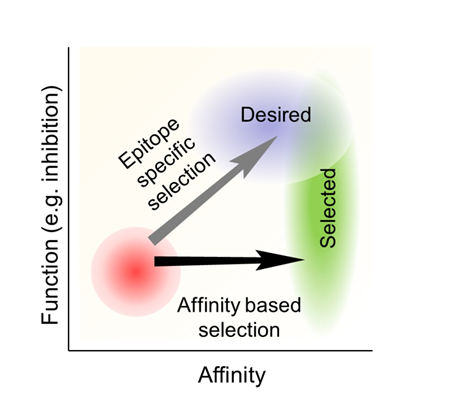
Figure 1: Inhibitors are often engineered by first optimizing the binding affinity. However, binding does not always correlate with function, and the engineered high affinity binders are screened further to identify the subset of the binders with desired function. By maximizing the affinity first, low affinity molecules are usually excluded from the final selection, even though many effective inhibitors may have low to medium affinity. A screen based on binding to a functionally important surface patch is more likely to simultaneously optimize function and affinity.
The success of a target neutral selection strategy depends critically on the starting library size, which is one of the most critical parameters that influence the ultimate outcome of the study.
In this short review, we examine the conceptual similarity between fragment based drug design (FBDD) and epitope guided protein inhibitor design. The conceptual overlap between the techniques distinguishes them from the HTS screens and the typical directed evolution studies, respectively. By outlining the differences between biased and unbiased search strategies, we highlight the potential benefits of explicitly incorporating biochemical and structural knowledge during inhibitor design. Based on this comparison, we speculate how the recent work on the engineering of cross reactive flu vaccine could have benefited from a systematic search for epitope specific binders.
Up and down strategies for engineering small molecular inhibitors
Although a number of small molecule enzyme inhibitors have been developed for therapeutic applications, the successful design of small molecule inhibitors of PPI remains challenging. The search for small molecule PPI inhibitors is often based on HTS of diversity oriented compound libraries (Figure 2a).

Figure 2: A) Screening a diversity oriented chemical library may yield a compound that binds the target protein with reasonable affinity, but the details of interaction are not sufficiently understood to allow efficient improvement of the interaction.
B) Fragment based drug discovery (FBDD) first identifies small molecules that bind the target at nearby locations and combines them into a larger molecule. The strength of interaction between the target and each fragment can be characterized and improved independently.
Because assembling and maintaining a large compound library is a significant undertaking, these experiments are usually conducted by pharmaceutical companies and large labs. To design a small molecule PPI inhibitor, the individual compounds in a library need be evaluated using a binding assay, such as fluorescence polarization spectroscopy or surface plasmon resonance [14,15]. Since binding to the target protein does not indicate functional inhibition, the selected binders need to be further screened in a secondary functional assay. Alternatively, a functional assay may be designed to screen the compounds based on their biological activity. The compounds that are identified from these screens often need to be characterized extensively and optimized.
Instead of the top down approach to drug discovery that is driven by automation and the availability of a large diversity library, compounds with desired binding properties may be designed rationally using computational and experimental techniques. Computational drug design includes simulated docking of potential compounds on the targeted surface to find those molecules that are likely to bind to the surface and thus have biological effects [16]. The binding affinity is predicted by summing over different energetic contributions, including van der Waals, electrostatic, and hydrogen bonding interactions. The entropic contributions to the free energy of binding are difficult to estimate accurately and account for large uncertainties in the prediction. Because allowing the target surface to move during the search can significantly increase the computational cost, the binding surface is typically modeled as a single structure (or a small number of related structures) [17]. Although water molecules play an important role in intermolecular binding [18], including them either during simulated docking or during molecular dynamics simulation also increases the complexity of calculation. Despite these limitations, computation can be useful in problems where experimental details are not available, including membrane proteins.
Fragment based drug discovery offers an attractive alternative to the traditional HTS (Figure 2b). As the name suggests, a high affinity binder is assembled by joining smaller molecules that independently show affinity to the targeted surface [19]. For example, if two small compounds bind nearby sites on the surface of a target protein, chemically joining them may result in a compound with improved affinity. Because the energetic contribution from each component is evaluated separately and can be individually optimized the molecular properties of the designed compounds can be understood in terms of the constituent components. The identification of lead compounds can be performed many different ways, including affinity measurements, crystallography, and NMR spectroscopy. Alternatively, the library compounds may be directed to the site of interest by using a natural ligand. FBDD is being used by several start-up companies engaged in drug discovery [20]. Numerous reviews have been written on the subject in recent years [21,22].
An important feature that distinguishes FBDD from high throughput screens is that the interaction is biased toward a specific surface patch that is known to be functionally important. The binding of potential molecular fragments are validated through various experimental steps, including biochemical and structural techniques, before they are assembled to a larger molecule. By focusing on the identification of small molecules that bind the targeted protein interface, FBDD systematically integrates relevant molecular information to design selective, high affinity inhibitors. Although the iterative nature of the process may appear to slow the discovery process, incorporating detailed interactions during the construction of molecular building blocks is thought to enable the ultimate discovery of higher affinity binders that work through a predetermined mechanism.
Imposing functional constraints during directed evolution of protein binders
The procedure for designing novel binders against a protein target is well established. There are a number of directed evolution platforms that can be used for this purpose, including phage, cellular (yeast, bacterial, and mammalian), and in vitro (mRNA and ribosomal) display systems [23]. The binders can also be engineered in various protein scaffolds. For example, in addition to the antibody scaffold (and the variants thereof), there are other well characterized scaffolds of different folds and tertiary structures, including β sheet, β barrel, α helical bundle, repeat motif, and other smaller proteins as well as short unstructured peptides. In some cases, the binding affinity can be engineered directly into a native protein (e.g. GFP), in which sequence diversity is introduced into a region of the protein that tolerates structural perturbation [24]. Different combinations of the template structure, target molecules, and the selection schema create opportunities for using engineered protein inhibitors to regulate biological processes that require specific intermolecular interaction.
There are challenges when developing engineered protein binders into useful therapeutics. Some of the challenges are biological. If the details of a biological process are not sufficiently understood at a molecular level, an engineered inhibitor, albeit of high specificity and affinity, may have little therapeutic benefits. Other challenges are technical. For example, preparing the target molecule for a directed evolution study may be difficult if the protein is unstable. The standard implementation of high throughput protein engineering, i.e. directed evolution, shares many features with the HTS employed for small molecule discovery. Importantly, the screen neglects biochemical and structural information available for the target. A custom built protein library may be used in the screen to reflect unique features of the target molecule, similarly to the way the composition of a small molecule library may be customized depending on the target molecule. This is often the extent to which the information about the target molecule is utilized to bias the selection.
Yet, by the time a protein engineer gets involved in a project (or a project takes on an aspect of protein engineering), there is already an abundance of biochemical and biological data on the targeted molecule. This creates opportunities to more efficiently “direct” the “evolution” of the binders. For example, mutagenesis studies may have been performed on the target molecules to show which residues are functionally important. The structure of the target molecule may also be available. It is not unusual that extensive structure-function study results are available through a multitude of studies. While these studies may have motivated the search for novel binders with potential inhibitory or stimulatory, effects on the molecule, the lessons from preceding studies are rarely incorporated during the engineering of a binder, and the engineering phase of the study is often treated independently of the preceding functional studies.
The disconnect between the functional studies on the target molecule and the engineering of epitope specific binders represents a less than ideal use of available knowledge. Since there is already functional data on the molecule, utilizing the information should improve the odds of discovering functionally relevant binders. It is true that a functional assay needs to be developed afterwards to test and confirm the engineered molecules regardless of whether the binders were engineered with or without being subjected to additional constraints. However, the protein binders that are engineered to target a defined surface patch only need to be confirmed and tested. On the other hand, the binders that are engineered without such constraints need to be further screened in a large scale functional assay. Depending on the functional assay that is needed, it may be difficult to develop an efficient functional screen for a large number of candidate molecules.
For small molecule design, FBDD provides a solution to how to incorporate existing biochemical and structural data during the inhibitor design. For directed evolution of protein binders, epitope guided engineering can be utilized to bias the selection based on available information (Figure 3).
Figure 3: Sorting a protein library based on binding to a target protein may result in a selection of clones that each binds at different locations. Although the affinity and selectivity of such interaction may be optimized, the screen does not optimize function of the engineered binders. On the other hand, a combination of positive (i.e. binding) and negative (i.e. no binding) selection using wild type and mutant target proteins can be used to identify the clones that are epitope specific. Epitope-guided engineering of protein binders thus yields clones that have desired physical, chemical and functional properties.
Importantly, epitope guided engineering allows constraints to be imposed to bias a functional selection without transforming the entire assay into a functional screen. That is, we can impose constraints that increase the odds (perhaps exponentially) of finding binders that are functionally relevant, but do so using a metric that is commonly used to design a binder. Since structure determines function, a lot of functional engineering can be done through the lens of structural engineering. If the structural properties of a binder can be carefully controlled, its function may follow. The benefit of taking this approach is that, at least for engineering binders against a structurally and functionally well characterized target, dictating the structural properties of the binder during affinity maturation, which can be relatively straightforward, may lead to binders with well-defined functional properties. In the process, one would have engineered a functional binder based on its structural properties and avoided the use of a large scale functional screen, which may be technically more challenging to implement.
We recently tested if functional constraints can be introduced during directed evolution by screening a monobody library in order to selectively bind a surface patch with a known function [25]. To this end, the binding of an engineered monobody to the conserved domain (CD) on the surface of Erk-2 should inhibit its function by preventing its association with substrates. Because this is already known based on biochemical and structural studies [26,27], targeting the CD domain should be equivalent to inhibiting its function. As expected, many of the engineered binders had expected inhibitory effect on Erk-2 and prevented its kinase activity. Therefore, the study demonstrates that by specifically targeting a functionally important site, we can predictably design molecules with defined function.
How cross-reacting, or universal, flu vaccine was discovered
Although it is obvious that an inhibitor would be most effective if it binds to a functionally important site, designing such inhibitors predictably is difficult. To this end, we consider the challenges of developing universal vaccines against the influenza virus. According to the world health organization, 5 – 15% of the world population is infected by influenza during a typical seasonal outbreak and nearly half a million infected individuals succumb to it annually [28]. Antibody based influenza vaccine can either prevent infection, if administered prophylactically, or provide therapeutic relief, if dosed after infection. Given uncertainties of the influenza strain that becomes active in any given year and the burden of stockpiling multiple antibodies, it would be highly desirable to design cross reacting, the so called universal, antibodies that are active against a large number of influenza strains.
There are two major groups and 16 subtypes of influenza viruses. Because the surface antigens targeted by influenza antibodies are highly variable among different subtypes, cross reactivity of an antibody raised against one subtype with another subtype is limited. That is, the antibodies are subtype specific and typically do not have broad neutralizing power. The reason for their subtype specificity is in part due to the way these antibodies interact with their targets. Antibodies typically bind their targets using a large surface area and the epitope of a therapeutic influenza antibody often includes both conserved and variable regions. This makes their interaction susceptible to sequence variation among different viral subtypes. Yet, there are also sites on haemagglutinin (HA) that are structural and functionally conserved. Not surprisingly, these are the sites that are targeted by known broadly neutralizing antibodies, including C179 [29] and C05 [30] (Figure 4a).
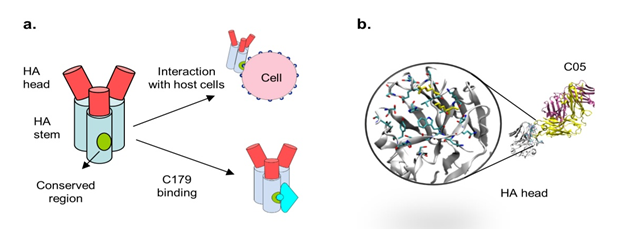
Figure 4 A) The influenza vaccine C179 binds a conserved region on the HA stem and is capable of neutralizing viruses expressing a broad range of HA subtypes, including H1, H2, H5, H6, and H9.
B) The cross reactive human antibody, C05, binds the HA1 head at a site that is conserved among different influenza subtypes. As such, C05 offers protection against different strains of influenza
The discovery of cross neutralizing antibodies is difficult to systematize, which requires identifying antibodies that bind to a structuraly and functionally conserved patch only. Taking the lessons from in vitro protein engineering studies, one way to engineer cross reacting antibodies would involve coordinated use of wild type and mutant HA stem proteins containing a targeted mutation within the site that is recognized by C05. For example, a small to large surface mutation, such as T155K or S193E, within the conserved region of the HA stem should abrogate binding at the conserved epitope and can be used to identify potential cross reacting antibodies (Figure 4b). The human antibody repertoire has been successfully reconstituted in an in vitro system, which should allow efficient selection of desired antibodies [31,32]. Importantly, the efficacy of an antibody depends on other parameters besides the affinity of binding, and may have therapeutic effects even when its affinity is low. Therefore, designing experiments to identify epitope specific antibodies may be far more important than optimizing the affinity of interaction.
Controlling the binding sites of a potential antibody can have a significant impact on human health. More than 30 million adults and children live with the human immunodeficiency virus (HIV). Currently, there is no cure and the availability of antiviral agents is limited in many parts of the world. The HIV envelope protein (Env) is a glycoprotein that is cleaved in the Golgi to gp120 and gp41, which then assemble to a trimer of heterodimers and mediate cell membrane fusion by binding to the CD4 receptor. Although the atomic structure of the Env trimer is not available, a useful model exists to locate the receptor binding site. The modeled structure may then be used to design a binding site mutant that can be used to engineer specificity of interaction [33]. For example, an antibody may be engineered to derive most of its binding energy from contacts to the conserved region. The antibodies that target a conserved and functionally important site on the viral surface should have inhibitory effects on viral entry.
CONCLUSION
In this review, we examined how small molecule and protein inhibitors may be engineered by biasing their interaction toward functionally relevant sites. Epitope guided engineering is underexplored in protein engineering but can be useful to identify binders with desired functionality.
ACKNOWLEDGEMENTS
The current work was supported by NSF CAREER (1053608) to SP.
REFERENCES
11.Wittrup WD. Protein engineering by cell-surface display. Curr Opin Biotechnol. 2001; 12:395-399.
20.Erlanson DA. Introduction to fragment-based drug discovery. Top Curr Chem. 2012; 317: 1-32.
22.Hajduk PJ. Puzzling through fragment-based drug design. Nat Chem Biol. 2006; 2: 658-9.
23.Park SJ, Cochran JR, Eds. Protein Engineering and Design. (CRC Press, ed. 1st, 2009), 1st.









































































































































{kind=link}