Hb Wroclaw (HBA1: c.266C>A) Detected by Capillary Electrophoresis Co-Inherited with Hb Icaria (HBA2: c.427T>A) Produces Erythrocytosis in an Italian Male
- 1. Laboratorio Genetica, Fondazione Ca’Granda, Ospedale Maggiore Policlinico, Italy
- 2. Laboratorio di Patologia Clinica Polispecialistica BIOS, Crotone, Italy
- 3. Sebia-Italia S.r.l. Bagno a Ripoli (FI), Italy
- 4. Formerly, U.O.C. Medicina di Laboratorio, Ospedale di Treviso, ULSS2 “Marca trevigiana” Treviso, Italy
- 5. Formerly, Laboratorio Genetica Umana, Ospedali Galliera, Genova, Italy
Abstract
Objectives: To date, more than 600 different α-globin gene defects, structural variants, and thalassemias have been described [1]. Thalassemias are mainly produced by greater or lesser extensive deletions or, more rarely, point defects. A variety of phenotypes can be observed when several thalassemia defects are inherited in combination with numerous Hb variants.
Methods: A 22-year-old man from Crotone (Calabria, Italy) with significant erythrocytosis was examined for hemoglobin (Hb) defects by capillary electrophoresis (CE). Molecular characterization was performed by direct sequencing of HBA1 and HBA2 genes and by multiple ligation-dependent probe amplification (MLPA) to detect deletions/duplications within the α-gene cluster.
Results: Two abnormal peaks were observed by CE: a main one in anodal position with respect to Hb A and a secondary one in anodal position with respect to Hb A2. Direct sequencing of HBA1 and HBA2 genes showed the presence of two point mutations respectively: HBA1: c.266C>A (Hb Wroclaw) and HBA2: c.427T>A (Hb Icaria, thalassemic variant).
Conclusions: Hb Wroclaw is a rare variant reported in HbVar in 2008 but not described in the literature. The mutation at position F9, codon 88 of the amino acid alanine (Ala), leads to substitution with glutamic acid (Glu). This mutation results in a structural change in the α-globin chain and, in the presence of Hb Icaria, produces a relative increase in Hb Wroclaw, this causes significant erythrocytosis in the proband and mother.
CITATION
Curcio C, Scarfò G, Pugliese L, Maoggi S, Barberio G, et al. (2023) Hb Wroclaw (HBA1: c.266C>A) Detected by Capillary Electrophoresis Co-Inherited with Hb Icaria (HBA2: c.427T>A) Produces Erythrocytosis in an Italian Male. Int J Rare Dis Orph Drugs 6(1): 1016.
INTRODUCTION
To date, more than 600 different defects in the HBA2 and HBA1 globin genes have been described [1]. These defects include thalassemias due to deletions and structural defects produced by mutations that partially or completely inactivate globin chain synthesis and manifest thalassemic behavior, and variants that are mildly symptomatic or totally asymptomatic especially if they affect only one of the four α-genes.
α-thalassemias (α-thal) occur in more than 120 different forms and are probably the most prevalent thalassemic conditions in the world population. This claim is based on the fact that many of these defects, when involving only one alpha globin gene, frequently present in clinically silent forms and may go undiagnosed. Mild forms of α-thalassemia (α+-thal) that lack only one of the four genes (-α/αα) responsible for chain synthesis are prevalent in Africa and Mediterranean regions, where homozygous forms (-α/-α) are also frequently found. Defects due to two nonfunctioning α-genes on the same chromosome (α0 thal) are prevalent in Southeast Asia. These genetic conditions are usually clinically asymptomatic, and individuals who carry them exhibit mild or moderate microcytosis with normal or slightly reduced Hb A2 levels. When three (-α/--) or all four genes (--/--) on the two chromosomes are missing or not functioning, clinical phenotypes classified as intermediate or severe thalassemias or “α-thal major” occur, as in the case of Hb H disease and Hb Bart’s hydrops fetalis syndrome, respectively [2-4]. In addition to the deletional α-thalassemias, there are more than 80 different defects in the HBA1 and HBA2 globin genes which are due instead to mutations involving one or a few nucleotides, and are still capable of producing thalassemic conditions. In these cases, the mutations mainly affect the regulatory steps of the gene (mRNA processing, translation, protein stability) and result in a reduced or complete inability to synthesize the α-chain. These defects are usually referred to as non-deletional α-thalassemia (αND-thal, with ND indicating non-deletion) and usually produce a more pronounced phenotypes when present in the homozygous state or associated with α0-thal defects. In fact, clinical phenotype are more severe when Hb H disease involves αND-thal defects compared with α+-thal deletion forms. [5]. Finally, there is the group of mildly symptomatic α-globin variants, which includes unstable variants and variants with altered affinity for oxygen (O2 ). In the latter case, the extent of conformational changes is not such that their function as oxygen transporters is significantly impaired [6]. Unstable α-variants are rarely associated with severe hemolytic anemia, and more commonly manifest their instability with thalassemia-like phenotypes [7]. In general, unstable α-variants or those with altered O2 binding, if inherited in the heterozygous state, rarely express marked or symptomatic phenotypes. As a result, such defects are infrequently detected, and poorly studied and documented in the literature. Among the α-variants that have an O2 binding defect, only 22 with increased affinity have been classified to date. In most cases they have mildly altered phenotypes, with minimal or undetectable secondary erythrocytosis. The more than 80 Hb β-variants with higher affinity for O2 normally have more pronounced phenotypes with detectable compensatory secondary erythrocytosis [1,8]. Erythrocytosis may be observed in α-variants, but is seen less frequently since the condition typically involves less than 25% of the α-chains produced, as it is usually present in only one of the four α-genes. In heterozygous β-variants, one of the two β-genes is involved, and thus the proportional presentation of abnormal β-chains will be much higher(~50%). Patients with an α-chain variant may approach the presentation of a β-variant only if the is inherited in the homozygous state or in combination with an α-thalassemia [9]. Finally, the relative amount of abnormal Hb of the α-chains will be greater when encoded by the HBA2 gene than when encoded by the HBA1 gene, and thus an identical mutation on different α-genes may lead to a different clinical presentation [10]. Erythrocytosis is a blood disorder characterized by an increase in the mass of red blood cells. The most common causes of erythrocytosis are acquired and caused by diseases and conditions accompanied by hypoxemia. This is what occurs in some variants of Hb in fact, due to increased affinity for oxygen, some degree of anoxia may occur in the tissues which is a stimulus for erythropoiesis resulting in erythrocytosis. The term erythrocytosis is sometimes used in association with or as an alternative to the term polycythemia, which, however, may have a broader meaning, to generally define those hematologic conditions characterized by an increase in hematocrit (PCV) and/or hemoglobin (Hb) concentration. It is rare for the clinician, observing a patient with erythrocytosis, to think of the presence of hemoglobinopathy as the first cause. Therefore, when faced with such evidence, it is always necessary to proceed with differential examinations [11]. Among these, measurement of partial pressure of oxygen at 50% saturation (P50) has been found to be sufficiently discriminating to prove or rule out the presence of a hemoglobinopathy. However, this type of measurement can produce false negatives, especially in the presence of α-globin defects or because of the preanalytical or analytical procedures associated with the test. In particular, false negatives can be caused by the slight deviations from the norm produced by some mutations, but especially by inaccuracy in the measurement of the P50 value [12]. In any case, the search for possible Hb structural defects in the presence of erythrocytosis is also appropriate and guidelines for the detection of hemoglobinopathies always recommend separation and relative quantification of Hb fractions with automated systems. Even then, both capillary electrophoresis (CE) and high resolution liquid chromatography (HPLC) may fail to recognize the presence of a hemoglobinopathy, so their combined use, as a first diagnostic step, is increasingly recommended to increase their discriminatory ability. Their results, when the presence of a hemoglobinopathy is suspected, should always be confirmed as part of a second diagnostic phase by direct study of globin genes. This approach is considered essential for adequate characterization of initially detected Hb variants and in the search for possible ‘silent’ defects, i.e., those Hb variants undetectable by such first-level analytical methods [13]. It should be added that it is important to diagnose and characterize Hb variants when there is suspicion of increased affinity for oxygen because, although this condition is usually well tolerated in a young patient, it can lead to thrombotic complications in elderly patients [14].
MATERIALS AND METHODS
Subjects
Three family members were enrolled in this study: the proband, the mother, and the father. Blood samples were taken from the proband for first level examinations and molecular analysis. The parents were only available for blood count. The study participants gave informed consent for the tests performed. All procedures were in accordance with the Declaration of Helsinki. The proband, a 23-year-old Italian man, presented to the laboratory with a request to evaluate an erythrocytosis also found in his mother in the past. The proband, who is in good health and does not engage in particular sports activities, denied smoking.
First level tests
Peripheral blood samples were collected in tubes containing tri-potassium salt of ethylenediaminetetraacetic acid (K3 EDTA) for analysis of erythrocyte parameters and Hb analysis. All assays were performed according to the manufacturers’ instructions. The blood count parameters were evaluated with the Automated Hematology Analyzer - BC-5380 - Mindray Global (High-tech Industrial Park, Nanshan, Shenzhen 518057, P.R. China). The separation and quantification of the hemoglobin fractions was performed by capillary electrophoresis (CE) (Capillarys 3 Tera kit with Hemoglobin(e); Sebia - Lisses, France). To assess the Hb stability, Carrell’s isopropanol test [15], and the search for erythrocyte inclusions in red blood cells after incubation with brilliant cresyl blue (BCB) was performed [16].
Molecular analysis
Genetic analysis was performed by extracting DNA from the leukocytes in the blood in K3 EDTA using an automated DNA extractor (QIAsymphonyTM; Qiagen GmbH, Hilden, Germany), followed by multiplex ligation-dependent probe amplification (MLPA) to detect deletions/duplications within the α-gene cluster, using the P140 HBA probe set (MRC-Holland, Amsterdam, The Netherlands) according to the manufacturer’s instructions. Sequencing of HBA2 and HBA1 genes was performed by Sanger sequencing (BigDye Terminator Cycle Sequencing Ready Reaction Kit v.1.1) on the ABI PRISMTM 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) [17].
RESULTS
The RBC indices of the proband and parents are summarized in Table 1.
Table 1: Main laboratory parameters of the proband and his parents
|
Parameter |
Unit |
|
Result |
|
Ref. range |
|
|
|
Proband |
Mother |
Father |
|
|
RBC |
1012/L |
6.95 |
6.43 |
5.40 |
4.50 - 5.90 |
|
HGB |
g/L |
172 |
158 |
158 |
13.0 - 18.0 |
|
PCV |
L/L |
0.54 |
0.51 |
0.47 |
0.37 - 0.49 |
|
MCV |
fL |
78.6 |
80.0 |
87.5 |
80.0 - 100.0 |
|
MCH |
pg |
24.7 |
24.6 |
29.2 |
26.0 - 34.0 |
|
PLT |
109/L |
86 |
160 |
181 |
150 - 400 |
|
Indirect Bilirubin |
mg/dL |
1.70 |
ND |
0.5 |
0 - 0.9 |
|
Ferritin |
g/L |
137 |
220 |
263 |
20 - 250 |
|
Hb A2 |
% |
1.3 |
NT |
NT |
2.5 - 3.2 |
|
Wroclaw Hb A2 |
% |
0.5 |
NT |
NT |
- |
|
Hb Wroclaw |
% |
28.0 |
NT |
NT |
- |
RBC: Red Blood Cells; HGB: Hemoglobin; PCV: Packed Cell Volume; MCV: Mean Corpuscular Volume; MCH: Mean Corpuscular Hemoglobin; PLT: Platelets; NT: not tested
Specifically, the proband and mother had increased PCV and RBC while MCH and MCV were at the lower limits of the normal range. The father had no alterations in erythrocyte indices. On CE examination the presence of an Hb variant in the “Z11” zone that was hypothesized to be produced by an α-gene defect, a minor peak in the “Z(S)” zone also being visible (Figure 1).
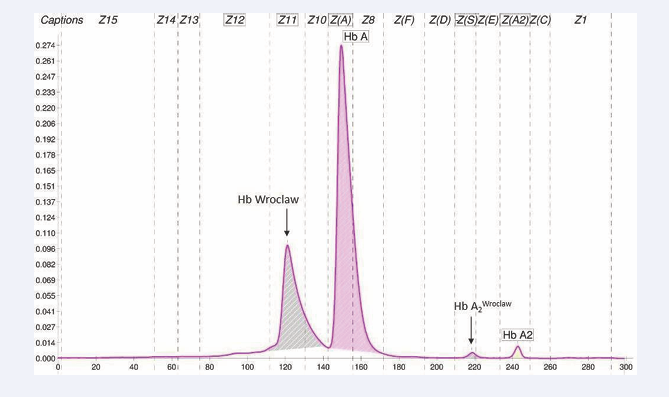
Figure 1: Capillary electrophoresis profile by Capillarys 3 Tera kit with Hemoglobin(e) of the proband with Hb Wroclaw in ‘Z11’zone.
The significantly reduced value of Hb A2 (1.3%) was also consistent with the additional presence of mutated Hb A2-X (0.5%) due to an α-variant, while the relative amount of variant Hb (28%) in the primary peack was significantly higher than the expected value for an α-variant. In the parents, analysis by CE could not be performed. Carrell’s test, performed on the proband’s blood, was negative, as was the search for intra-erythrocyte inclusions of Hb H like or Heinz bodies after incubation with brilliant cresyl blue at 37°C.
MLPA of the α-globin gene cluster did not reveal the presence of any deletional defect. Direct sequencing of the α-globin genes identified a heterozygous point mutation HBA1: c.266C>A; [88(F9) Ala>Gluα (Figure 2A)
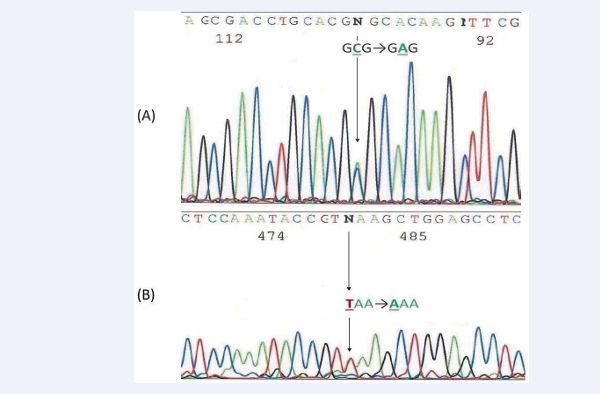
Figure 2 A: Part of DNA sequencing of the HBA1 gene showing the substitution of a single nucleotide (C>A) at codon 88 of exon 2 (Hb Wroclaw); 2B: Part of DNA sequencing of the HBA2 gene showing the substitution of a single nucleotide (T>A) at stop codon 142 (Hb Icaria)
corresponding to the Hb Wroclaw variant [1] and a heterozygous point mutation HBA2: c.427T>A; 142Stop>Lys (Figure 2B) corresponding to the Hb Icaria, a nondeletion α-thalassemia mutations [18]. This is an elongated and unstable variant of Hb with a modified C-terminal sequence: (142)Lys-Ala-Gly-Ala-Ser-Val-Ala-Val-Pro-Pro-Ala-Arg-Trp-Ala- Ser-Gln-Arg-Ala-Leu-Pro-Ser-Leu-His-Arg-Pro-Phe-Leu-Val Phe-(172)Glu-COOH.
DISCUSSIONS
Hb Wroclaw is a rare variant of alpha globin chains reported by Mantio D. et al., in HbVar in 2008 [1], observed in a Polish subject and not described in the literature. The mutation on the HBA1 gene leads to the replacement of an alanine residue (Ala) with a glutamic acid residue (Glu), at position F9, near the end of the F helix, at codon 88. The Glu amino acid residue is slightly larger than the Ala residue it replaces, has hydrophilic side chains and acidic characteristics. In addition, residue F9 is adjacent to histidine residue (His) F8 proximal to the α-chain. This proximity may condition the shifts of the His residue during coordinated configurational changes that binding and oxygen release [19]. The result, in this case, is a higher affinity for O2 resulting in a bone marrow response and consequently a more pronounced erythrocytosis, probably also due to the co-inheritance of Hb Icaria, a point mutation variant with thalassemic characteristics. This case presents phenotypic features similar to the five other variants described in the literature with the Ala residue differently substituted at codon 88 of the alpha globin chains (Table 2).
Table 2: Known variants of alpha globin chains due to a mutation at codon 88 (F9).
|
|
Hb name |
HGVS name (a) |
Hb X % |
O2 Affinity |
Erythrocytosis |
Refs. |
|
1 2 |
Hb Wroclaw a 88(F9)Ala>Glu Hb Wroclaw a 88(F9)Ala>Glu |
HBA2:c.266C>A (or HBA1) HBA1:c.266C>A |
21.7 28.0 |
- - |
Unknown Yes |
[1] (b) |
|
3 |
Hb Loire a 88(F9)Ala>Ser |
HBA2:c.265G>T (or HBA1) |
25.0 |
Increased |
Unknown |
[20] |
|
4 |
Hb Columbia-Missouri a 88(F9)Ala>Val |
HBA2:c.266C>T (or HBA1) |
- |
Increased |
Yes |
[19] |
|
5 |
Hb Valparaiso a 88(F9)Ala>Gly |
HBA2:c.266C>G (or HBA1) |
27.0 |
Increased |
Unknown |
[21] |
|
6 |
Hb Voorhees a 88(F9)Ala>Thr |
HBA2:c.429A>T |
16.5 |
Increased |
Yes |
[1] |
(a) The mutated a-gene has not been characterized and is not described in the literature for Hb variants 1,3,4,5; (b): see text
The information available in the literature does not seem sufficient for these Hb variants to exclude the presence of co-inherited α-thal defects [1,19-21]. We consider it important to specify the type of mutated gene at codon 88 related to the variants shown in Table 2. As previously mentioned, it is known that the same mutations can be expressed differently on different genes (HBA2 or HBA1) that we know to be paralogous [22]. In fact, these genes share a high degree of similarity in DNA sequence, producing identical α-globin chains but with different synthetic efficiency. It is known that the average synthesized amount of an Hb variant produced by a mutation in the HBA2 gene is higher than that of an Hb variant produced by the same mutation in the HBA1 gene. It follows that the presence of variants in different relative amounts could correspond to higher phenotypic expression in the case of mutations in the HBA2 gene. In the case we studied, Hb Wroclaw, present on the HBA1 gene, was quantified as 28%. This percentage is higher than that found for α-variants on average and, in particular, for those on the HBA1 gene. However, we also ascertained in the proband the presence of the αND-thalassemic variant Hb Icaria. This is probably the cause of the quantitative increase in Hb Wroclaw that consequently produced detectable erythrocytosis. It has already been mentioned that qualitative and quantitative Hb tests by CE were not performed in the parents. The only relevant data available was the erythrocytosis present in the mother. Based on this observation we consider the probability that the proband’s mother may also have the two defects characterized in her son and they were transmitted in cis on the same chromosome. However, this would need to be proven via further characterization tests that could not be performed on this occasion. As for Hb Icaria, which has been described mainly in European subjects, it has very similar phenotypic features to the other six variants of the HBA2 gene that have a mutated stop codon at position 142 (Table 3).
Table 3: Some characteristics of the six thalassemia variants that have an elongated alpha globin chain of 31amino acids as a result of a mutation at the TAA (142) stop codon
|
|
Hb name |
HGVS name |
Hb X, % |
CE zone |
MCV, fL |
Refs. |
|
1 |
Hb Icaria a2 142, Stop>Lys |
HBA2:c.427T>A |
- |
- |
75-80 |
[18] |
|
2 |
Hb Constant Spring a2 142, Stop>Gln |
HBA2:c.427T>C |
1-3 |
Z(C) |
61-87 |
[23] |
|
3 |
Hb Koia Dora a2 142, Stop>Ser |
HBA2:c.428A>C |
<2 |
Z(C) |
75-85 |
[24] |
|
4 |
Hb Seal Rock a2 142, Stop>Glu |
HBA2:c.427T>G |
3-4 |
Z(E) |
70-75 |
[25] |
|
5 |
Hb Pakse a2 142, Stop>Tyr |
HBA2:c.429A>T |
<2 |
Z(C) |
80-85 |
[26] |
|
6 |
Hb Kinshasa a2 142, Stop>Leu |
HBA2:c.428A>T |
- |
Z(C) |
82 |
[27] |
Hb X, relative values of each Hb variant obtained by different separation methods and taken from the literature. CE zone, the migration zones by capillary electrophoresis (the migration zones of variants 3-6 were assumed based on the characteristics of theamino acids replacing codon 142, while the information on Hb variants 1 and 2 was acquired by the authors). MCV, Mean Corpuscular Volume (values found in heterozygous subjects).
These have been described mainly, but not solely, in Asia. These defects, in association with α0-thal, produce particularly severe forms of Hb H disease [28,29]. The need for transfusions has been documented in many cases, especially in the early years of life, and very high percentages of Hb H have been observed. This shows that Hb Icaria, like the other Hbs shown in Table 3, expresses marked thalassemic characteristics [30]. Hb Icaria is also considered an unstable Hb,but this characteristic is not detectable by classical laboratory tests because the instability is so marked that it results in rapid post-translational denaturation of the globin molecule [31]. This instability is demonstrated in the current case by the total absence of peaks related to this variant (Figure1), even though the mutation associated with Hb Icaria produces a change in total electrical charge that would be consistent with a peak visible by CE. The other variants similar to Hb Icaria, shown in Table 3, are also difficult to detect by traditional instability testing, but are separated by CE and displayed in consistently small amounts [32]. Future functional characterization of Hb Wroclaw in the test subject and his parents might include measurement of P50 for better assessment and management of O2 saturation in the patient. A low P50, indicating a higher affinity for O2 , would be expected in accordance with the erythrocytosis and defects found. However, this was beyond the scope of the studies reported here, which were conducted with the aim of completing the molecular characterization of the HBA2 and HBA1 genes following the results of the first-level tests. Recent methodological guidelines recommend this approach whenever a structural and functional abnormality is found during routine clinical examinations [12, 33,34].
Finally, the study of this proband and his parents showed that hemoglobinopathies are characterized by extreme heterogeneity and multiplicity of defects and phenotypes. Sometimes this can make the laboratory’s task particularly complicated. However, the use of traditional tests alongside increasingly sensitive and specific tests and the use of updated diagnostic pathways may prove to be important requirements for a good characterization of these defects. In addition, information from patients and family history is always necessary for an appropriate approach to laboratory investigations and for proper genotype-phenotype correlation, supporting dedicated genetic counseling that today can also make use of evolving in silico tools [13,35,38].
REFERENCES
2. Higgs DR. The molecular basis of α-thalassemia. Cold Spring Harb Perspect Med. 2013; 3: a011718.
5. Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010; 5: 13.
31. Barberio G, Ivaldi G. Unstable hemoglobin variants: A challenge for the laboratory? Biochimica Clinica. 2022; 46: 96-116.








































































































































