A
- 1. Clinical Services, Clinical Research and Evaluation, USA
- 2. Clinical Services, Emerging Therapeutics, USA
- 3. Quality and Accreditation, Diplomat Pharmacy, USA
Abstract
Rigorously testing the growing medication pipeline for rare conditions continues to be an industry challenge. The low prevalence rates for rare diseases run contrary to the phased drug development processes that are the standard and poses significant obstacles for developing, testing, and approval of promising treatments. The intent of this position paper is to bring together into a practical framework three elements of clinical research (N-of-1, adaptive design techniques, and multiple baseline) for testing small and variable samples of patients undergoing treatment. The objective is to bring continuity and best practices to the evaluation of pharmaceutical products for treating rare disease. Specifically, the paper discusses general principles for when and how they should be applied and what limitations should be considered when developing methods for testing treatments for rare diseases.
KEYWORDS
• N-of-1
• Clinical trials
• Adaptive design
• Multiple baseline
• Niemann-Pick Type C
• Real world outcomes
CITATION
Schwartz S, Chandanais R, Yazdanfar M (2019) A “Small” Data Approach to Evaluating Clinical Outcomes in Rare Disease. Int J Rare Dis Orph Drugs 3(1): 1008.
PERSPECTIVE
Orphan drug trends
Rare diseases in the U.S. are defined as having a patient population of <200,000. Although each disease impacts a relatively small patient population, there are estimated to be approximately 7,000 documented rare diseases. This corresponds to about 25-30 million people in the U.S. alone managing a rare disease [1]. Considered in total, rare disease touches us all.
Historically, pharmaceutical manufacturers have shown little interest in developing rare disease treatments, in part due to a perceived limited economic potential relative to development costs. For example, between 1973 and 1983, fewer than 10 rare disease treatments reached the U.S. market. To encourage the development of drugs to treat rare diseases, the FDA Office of Orphan Products Development (OOPD) implemented several programs including the Orphan Drug Designation, Rare Pediatric Disease Designation, Priority Review Voucher, Humanitarian Use Device, and various grant programs to support greater innovation in this area [2].
As a result, the orphan disease drug pipeline has become one of the most active in drug development today. Since 1983, more than 600 agents treating rare diseases have reached the market. Additionally, there are more than 560 products in development for the treatment of rare diseases. Many manufacturers now see rare diseases as an opportunity to treat patients with an unmet medical need in a disease state where there is often little or no competition for market share. This has also been an inroad for smaller manufacturers to enter the market. Despite the increased interest and number of product approvals, about 95% of rare diseases still lack an FDA-approved treatment option [3].
Rigorously testing this growing medication pipeline for rare conditions continues to be an industry challenge. The low prevalence rates for rare diseases are contrary to the phased drug development processes that are the standard in the pharmaceutical industry today. Consequently, rare diseases pose a number of significant obstacles for developing, testing, and approval of promising treatments.
1. The range of methodological issues impacting drug development has been discussed in broad context elsewhere [4-7]. The intent of this position paper is to bring together, into a practical framework, three elements of clinical research for testing small and highly variable samples of patients undergoing treatment. The objective is to bring continuity and best practices to the evaluation of pharmaceutical products for treating rare disease. Specifically, this paper will cover three approaches, discuss general principles for when and how they should be applied, and discuss what limitations should be considered when developing methods for testing treatments for rare diseases. The paper will specifically address the following:N of 1 Trials. These trials are designed, conducted, and evaluated at the level of the individual patient or small group, with the intention of objectively optimizing treatment for an individual patient [8].
2. Adaptive Research Design. A defined trial allows for prospective modifications to the design based on accumulating data feedback on the trial subjects [9,10].
3. Multiple Baselines. The multiple baseline design staggers the length of baseline and onset of intervention with repeated measures across treatment conditions such that each consecutive individual serves as both control and treatment subject [11].
N-of-1 Methodology
Aggregated data commonly used in randomized clinical trials (RCTs) and, more recently, in big data approaches use deductive inference that works from a forest view of clinical outcomes based on the greatest overall outcomes (i.e. group averages) generalized to a specific patient. This type of reasoning is both logically sound and has been associated with advances in clinical treatments that help the most people the most. For high-prevalence conditions, the power (or ability to detect a treatment effect) of testing a promising treatment is based in part on the size of the sample tested (i.e. more patients tested more power to detect a treatment effect). However, the RCT has more recently been subject to some criticisms primarily focused on its limitations as the “sole” source of evidence. The general acceptance of this model by the healthcare industry and regulatory institutions has ignored a complementary measurement and evaluation approach that makes greater use of inductive reasoning — namely, N-of-1 treatment designs and analysis.
The rigor and logic of N-of-1 designs have been well articulated and expanded upon for over a half century. Yet they have never achieved widespread adoption by practitioners or patients because the labor and time required for collecting the time series data necessary for N-of-1 analyses was prohibitive. However, with the growing availability of streaming data via digital, mobile, wearable, and other evolving technologies, this barrier is no longer an issue. By using more refined time-ordered data (data measured more frequently over time) to optimize evaluation of individual-level treatment response over time, proof points regarding both efficacy and effectiveness can be expedited (as well as identification of AEs). Such approaches are particularly well suited to the acquisition of real-world data. Additionally, N-of-1 approaches are truer to clinical practice by providing individualized evaluation and feedback to the patient and clinician about the quality and strength of their unique treatment response, which have been shown to enhance patient/ clinician dialogue [12]. Further, like more traditional approaches, N-of-1 can incorporate biological (genomic), behavioral, psychological, and digital health data. The N-of-1 framework does not challenge the more conventional group science or trendier big data but actually complements them with strategies around time-ordered data within a single individual and then aggregates across multiple individuals.
Adaptive design
Adaptive designs have become of greater interest and are part of the FDA’s approach to innovating and streamlining the drug development pipeline and approval process. Accordingly, the FDA defines adaptive studies as those which prospectively allow for possible planned study modifications based on study data and experience that have accrued up to some defined interim evaluation cadence [13]. Such changes can be procedural or analytical [11,14]. Such adaptation is an inherent part of the N-of-1 trial and is also much truer to clinical practice and decision-making, where either staying or changing the course of care is driven by the feedback of clinical data.
Multiple baseline
The multiple baseline design is one in which the start of treatment is staggered across individuals. Figure 1 graphically displays a basic multiple baseline design. In Figure 1, three baseline cohorts are defined by three (safe) baseline lengths (A Phase) to which patients can be randomized. Patients are randomized again, at the onset of the B Phase into an active treatment or comparison (control) condition. The staggering of treatment onset helps control for a host of other potentially unknown confounders (much in the way randomization does) and supports stronger causal conclusions when the clinical change (i.e. outcomes) observed is temporally contingent with the onset and course of treatment. C phases can constitute additional treatments, titration of the B Phase treatment, or, in a true reversal design, would be the treatment not assigned in the B Phase.
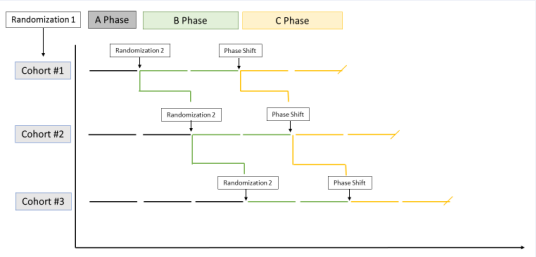
Figure 1: Basic Multiple Baseline Design.
An Example Use Case - Niemann-Pick (Type C)
Niemann-Pick type C (NPC) is a rare autosomal recessive disease affecting approximately 2,000-3,000 individuals globally. It is one of a family of lysosomal storage disorders that affects the ability of the body to metabolize cholesterol and other lipids. This diminished metabolization results in the accumulation of lipids within organs and tissues including the brain, liver, and lungs, ultimately resulting in death. NPC is caused by mutations of the NPC1 or NPC2 genes, with NPC1 mutation being the most common [15,16].
The onset of symptoms is highly variable (occurring along the developmental pathway from infancy to adulthood). Symptoms of NPC may include ataxia, vertical supranuclear gaze palsy, dystonia, liver disease, interstitial lung disease, difficulty speaking and swallowing, reduced mental function, and seizures. Individuals with Niemann-Pick type C generally begin exhibiting symptoms in childhood and may live to become adults. There is no FDA-approved treatment for NPC. However, miglustat is approved by the European Medicines Association (EMA), as well as the regulatory organizations of some other countries [15,16].
Treatment Pipeline
Recently, a pivotal randomized clinical trial to support FDA approval for a promising cyclodextrin compound known as adrabetadex or VTS-270 for the treatment of NPC failed [17]. Importantly, the study of 56 patients over 52 weeks failed in part because neither the treatment nor control condition demonstrated worsening of their clinical course, leaving researchers unable to determine exactly what the results mean. The negative results may have multiple explanations, some of which do not reflect on the efficacy of the medication itself. In a journalistic article reporting on the trial outcomes [17], the trial design was questioned. Importantly, without change (i.e. variability) in the primary outcome in either or both of the treatment and control groups, it is next to impossible to demonstrate a treatment effect. Interestingly, an earlier phase 1-2 trial concluded that VTS-270 slowed progression of NPC, as measured by improvement in certain neurological severity scores [18].
The results of these two studies seem to be at odds with each other and leave a great deal of uncertainty regarding the disease progression of NPC and the efficacy of adrabetadex. Alternative techniques for testing efficacy such as N-of-1, multiple baseline, and adaptive design techniques may have been useful in providing further insight into the natural course of NPC, as well as the safety and efficacy of potential treatments.
Application of Combined Methodology to an NPC Trial
In this fictionalized use-case design example, a promising NPC treatment will serve to illustrate how these approaches might work together. Figure 2 defines a 3 X 2 X 2 mixed model factorial design whereby consecutive qualifying subjects are initially randomized into one of three varying baseline periods. This consecutive recruitment and randomization can occur at any point in the drug development process (including the use of retrospective and prospective approaches to baseline), and the structure is friendly to post-market registries as well. At the end of the baseline phase (A), patients are randomized a second time to either of two treatment conditions (i.e. test article vs. a true placebo/standard of care). In the C Phase, subjects cross over to the treatment not assigned in Phase B. These two condition levels are conceptually like a more traditional RCT but prospectively consider each participant as both control and experimental subject. The crossover from B to C phase’s accounts for order or carryover effects and, in the case where the test article was assigned, this arm represents a true “reversal” or ABA design. At predetermined points, interim analyses are scheduled and conducted, with a priori decision rules regarding continuation and/or modification (e.g. dose change). The final phase is an “open label” phase, which is offered only if the final analysis supports a positive treatment effect and ongoing monitoring (assessing real-world outcomes and patient challenges/barriers/ issues) for a given individual patient.
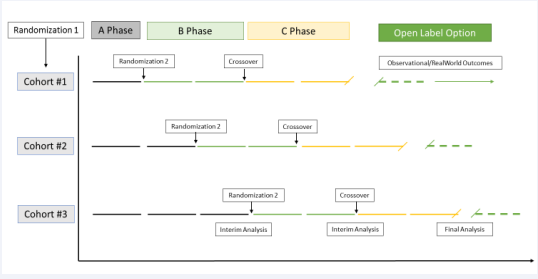
Figure 2: A Mock Pivotal Trial Multiple-Baseline Design
Abbreviations: NPC: Niemann-Picks Disease Type C.
• 3.8. Considerations and ConstraintsRate of clinical change. When considering the use of N of 1 and multiple baseline methods, the expected rate of change of the clinical condition and hypothesized treatment effects are important considerations (and informs decisions in the remainder of these considerations). Importantly, the expected rate or speed of change must be thought out when deciding upon a phase length (and any washout phases). Longer periods of change allow for the possible intrusion of confounding factors.
• Run in staggered baseline and clinical/ethical considerations. For conditions with an effective standard of care option (SOC), SOC can serve as the baseline. However, when treatment options are limited and the natural course of the disease is characterized by rapid decline; historical baselines can sometimes be employed to expedite initiation of the treatment phase. It is also a common misconception that the baseline phase must show stability (often interpreted to mean unchanging). N of 1 can accommodate trending baseline phases but is less robust when baseline data are erratic.
• Need for counterbalancing. Given that crossover elements are essential to the logic and efficiency of these designs, it is important to control for order or effects that may serve as confounds to interpretation of treatment effects in a given phase. Varying counterbalancing models such as Latin Square can be used, depending on the number and levels of the conditions being tested.
• Need for washout. When two (or more) active treatments are being compared, the crossover to the other treatment condition may first require a washout period based on the half-life of the medication(s) being tested.
CONCLUSIONS AND RECOMMENDATIONS
Rigorously evaluating medications intended for rare conditions continues to be a methodological and regulatory challenge for the industry. By definition, the low prevalence rates that characterize rare diseases undermine the power to detect treatment effects using traditional statistical approaches within the classic phased drug development processes. FDA guidance to perform nontraditional studies creates the ‘broadest flexibility” for evaluation, which is needed, but also leaves the industry with ambiguous direction. This includes absorption, distribution, metabolism and excretion (ADME), risks, toxicology, and unique study designs [FDA guidance doc].
In this article, we have presented three methodological design approaches that are not new but are infrequently used in clinical trials research and yet can add to the power and sensitivity of clinical efficacy testing when sample sizes are small (under powered by conventional statistical standards). Advancing a different perspective for a smaller number of individuals provides for the future of research and discovery in rare disease.
The approach described above aligns closely with the FDA guidance in the following ways:
1. This methodological approach employs an adaptive design whereby consecutive patients are enrolled and monitored such that early evidence informs subsequent phases from early safety testing through post-approval real-world outcomes monitoring.
2. Like more traditional approaches, N-of-1 can incorporate biological (genomic), behavioral, psychological, and digital health data such that users themselves can begin to evaluate the relationships of their own treatment response patterns and the contingencies that impact them.
3. Rigorous, statistically valid, natural history–controlled, cross-over, and n-of-1 trials can establish efficacy and support regulatory approval of new treatments for rare diseases [5]. This system accommodates traditional methodological controls for ensuring internal validity, including the two primary features of the classic RCT (i.e., randomization and blinding) while effectively optimizing the value of each patient as representing both control and treatment conditions (increasing power per patient and reducing inter-individual variability).
4. Substantial evidence: Thoughtful combination of the described design elements (when applied with consideration of the limitations) allows for strong causal conclusions when temporal contingency between onset of treatment and change in the primary (and/or secondary) outcomes with adequate power, comparison, and confound control.
5. N-of-1 approaches are truer to clinical practice than RCTs, by providing individualized feedback to each user (or clinician) about the quality and strength of their unique treatment response. For the clinician, this revitalized form of scientific and behavioral interaction evaluation can help them validate or reject the impact a given treatment has for a given patient with increased efficiency and accuracy [19].
REFERENCES
2. U.S. Food & Drug Administration. Developing Products for Rare Diseases and Conditions. 2019.
3. Pharmaceutical Research and Manufacturers of America. Rare Disease by the Numbers. 2019.
15. Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010; 5.
17. Wadman M. Drug for rare disease disappoints in key trial.








































































































































