A Case Report of Alport’s Syndrome: A Rare Cause of Anterior and Posterior Lenticonus
- 1. Ophthalmology Resident, Tishreen University Hospital, Syria
- 2. Master of Ophthalmology, Tishreen University Hospital, Syria
- 3. Professor & Head of the Ophthalmic Division, Tishreen University Hospital, Syria
Abstract
Background: Alport’s syndrome (AS) is a heterogeneous basement membrane disease characterized by hematuria with progressive hereditary nephritis, high-frequency sensorineural hearing loss (SNHL), and pathognomonic ocular lesions. The ocular finding of Alport’s syndrome has been reported only a handful of times previously in the literature. We described a case of Alport’s syndrome, its ophthalmic features, clinical manifestation, and diagnosis.
Case presentation: A 59-year-old female patient of Arabian descent presented blurred vision associated with progressive hearing loss. Her uncorrected distance visual acuity (OD: 0.01&OS: 0.06). Slit lamp examination of eyes revealed bilateral anterior and posterior lenticonus with retroillumination, oil droplet reflex seen through retro illumination. Audiometry revealed bilateral severe sensorineural hearing loss for higher frequencies. Renal ultrasound showed bilateral small kidneys, with poor cortico-medullary differentiation parenchymal changes consistent with nephritis. A urine examination revealed mild proteinuria with increased blood urea (151 mg/dl) and serum creatinine (2.60 mg/dl).
Conclusion: It is important to recognize Alport’s syndrome early in the course of the disease. This is facilitated by an integrated approach to diagnosis. The role of an ophthalmologist is prudent in the diagnosis of such interdisciplinary pathological entities. There should be a high index of suspicion for Alport’s syndrome in any patient presenting with anterior and posterior lenticonus. Early diagnosis can improve longevity and improve the prognosis of Alport’s syndrome patients.
Keywords
Alport’s syndrome, Lenticonus, Nephritis, Sensorineural deafness
CITATION
Qasem A, Karroum R, Darwish T (2023) A Case Report of Alport’s Syndrome: A Rare Cause of Anterior and Posterior Lenticonus. Int J Rare Dis Orph Drugs 6(1): 1013.
ABBREVIATIONS
AS: Alport’s Syndrome; SNHL: Sensorineural Hearing Loss; BMI: Body Mass Index; BPM: Beat per Minute; BP: Blood Pressure; CRF: Chronic Renal Failure; GBM: Glomerular Basement Membrane
BACKGROUND
AS described by Cecil A. Alport in 1927, Alport’s syndrome (AS) is an inherited disorder of many forms, most commonly X-linked. It typically presents with the classic triad of progressive glomerulonephritis, progressive high-tone hearing loss, and several ocular signs, the most pathognomonic of which is the presence of anterior lenticonus [1].
Anterior lenticonus is a rare developmental anomaly in which there is a spherical protrusion of the anterior surface of the lens into the anterior chamber, causing gradually progressive axial myopia. Most cases are associated with Alport’s syndrome. In contrast, posterior lenticonus, the bulging of the posterior surface of the lens into the vitreous, is not specific to AS. Anterior and posterior lenticonus in the same eye is very rare but may be present simultaneously in classical Alport’s syndrome [2,3]. We reported a case of Alport’s syndrome initially presenting with gradually progressive blurring of vision in both eyes associated with bilateral anterior and posterior lenticonus.
CASE PRESENTATION
A 59-year-old female patient of Arabian descent presented with blurred vision. Her history started when she was 18 yrs. old with a history of progressive hearing loss associated with blurring of vision with glare and abnormal head posture.
The blurring of vision worsens in the last 2 years with progression in severity; she does not note any problem related to near vision.
She had a history of colicky abdominal pain in her loins that started suddenly 4 yrs. ago, that radiated to her genitalia, the patient denied hematuria or any urine changes, and the episode was repeated approximately 3 times a year.
Her cousin had nephritis, hearing disturbance, and blurring of vision, and he underwent dialysis and died in his 25th yrs. of life.
Her nephew was diagnosed with nephritis that progressed to renal failure and he was on hemodialysis therapy for 5 yrs.
Her niece was diagnosed with nephritis that progressed to renal failure and she was on hemodialysis therapy for 4 yrs. then died in her 28th of life.
On examination, the patient looked pale, had a normal BMI, pulse rate of 74 BPM, and BP 140/85 mmHg.
The Cardiac exam was within normal, respiratory examination revealed normal vesicular breathing equal bilaterally, and the abdominopelvic exam was within normal with no lower limb edema.
Her uncorrected distance visual acuity (OD 0.01 and OS 0.06). Slit lamp examination of both eyes revealed bilateral anterior and posterior lenticonus, oil droplet reflex seen through retroillumination [Figure 1,2] with posterior subscapular cataract.
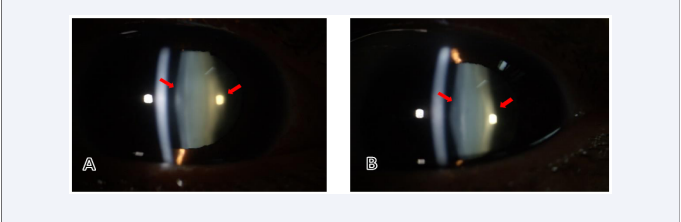
Figure 1: Slit-lamp examination of the right eye (A) and left eye (B) showing the posterior lenticonus (red arrows)..
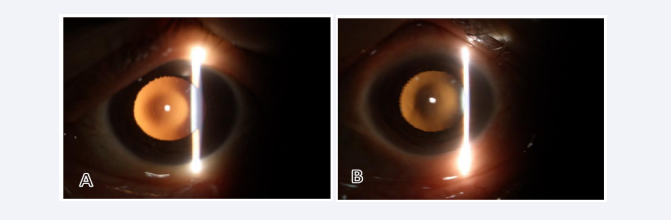
Figure 2: Oil-droplet reflex in right (A) and left (B) eyes
An anterior segment scan using a scheimpflug camera revealed an obvious Right & Left anterior and posterior lenticular bulge [Figure 3].
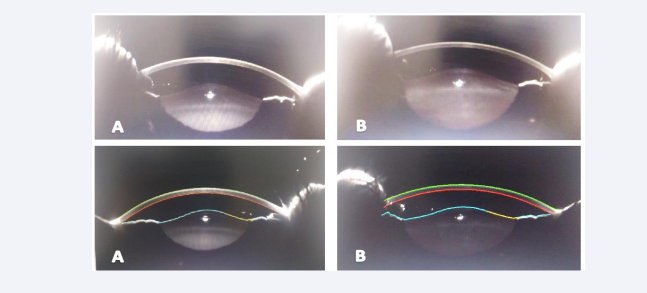
Figure 3: Anterior segment scan using a scheimpflug camera reveals Right (A) & Left (B) anterior and posterior lenticular bulges.
IOP by puff tonometer (OD 34 mmHg & OS 29 mmHg), Anterior chamber Gonioscopy showed open and normal angles, and C/D ratio (OD 0.7&OS 0.6).
Her investigations showed Hb 8.8 gm/dl, WBC count 6300/ mm3, increased blood urea
(151 mg/dl), serum creatinine (2.60 mg/dl), and electrolytes were within normal limits. Urine output for 24 hours was approximately 1800 ml/day and showed mild proteinuria. Renal ultrasound showed bilateral small kidneys (8.5 x 4.1 cm right, 7.5 x 3.2 cm left) [Figure 4] with poor cortico-medullary differentiation parenchymal changes consistent with nephritis. Audiometry revealed bilateral severe sensorineural hearing loss was observed at higher frequencies [Figure 5]. Invasive renal biopsy and genetic analysis were not performed because of financial constraints.
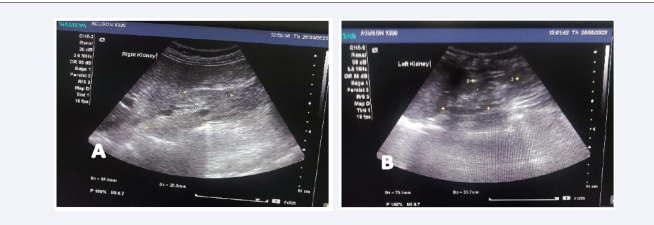
Figure 4: Transabdominal ultrasonography reveals Right (A) & Left (B) renal small size with poor corticomedullary differentiation.
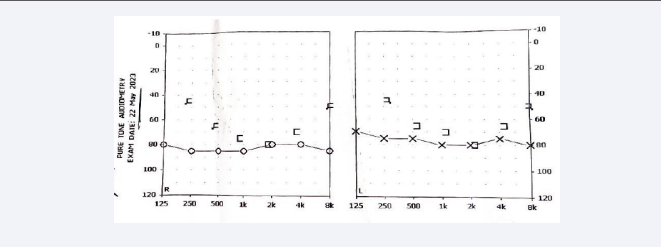
Figure 5: Audiometry reveals moderate sensory neural hearing loss..
Conclusions
It is important to recognize Alport’s syndrome early in the course of the disease. This is facilitated by an integrated approach to diagnosis. The role of an ophthalmologist is prudent in the diagnosis of such interdisciplinary pathological entities. There should be a high index of suspicion for Alport’s syndrome in any patient presenting with anterior and posterior lenticonus. Early diagnosis can improve longevity and improve the prognosis of Alport’s syndrome patients.
DISCUSSION
Alport’s syndrome (AS) is a progressive glomerulonephritis that is associated with high-tone sensorineural deafness and characteristic eye signs. It accounts for 0.6% of all patients who start renal replacement therapy in Europe and is most commonly inherited as an X-linked disorder with a gene frequency of 1 in 5000 [1]. During the last six years, several type IV collagen genes have been implicated in the etiology of AS, and mutation detection studies are enabling genotype/phenotype correlations to be made, as well as facilitating carrier detection and prenatal diagnosis [1].
The term Alport’s syndrome has been used extensively for patients with a variety of clinically heterogeneous hereditary nephritides, including some without deafness, and even benign familial hematuria. Only a few authors have used strict diagnostic criteria to define a clinically homogeneous subgroup of families with “classical” AS [4,5].
Classical (X-linked) Alport’s syndrome
In 1988, a set of four clinical diagnostic criteria was described that enable the identification of families which are affected with the same hereditary nephritis as Alport’s original family. “For a diagnosis of AS to be made clinically, any patient (or other affected relatives) with unexplained hematuria must fulfil at least three of the four criteria listed below (different features may occur in different subjects within the family):
(1) positive family history of macro/microscopic hematuria, chronic renal failure (CRF), or both; (2) electron microscopic evidence of Alport’s syndrome on renal biopsy; (3) characteristic ophthalmic signs, that is, anterior lenticonus or white macular flecks or both; and (4) high tone sensorineural deafness. Thus, an affected male with a negative family history may be diagnosed as having AS with confidence only if he has all the typical clinical signs. The clinical criteria are considered in more detail below.
(1) Positive family history
The importance of obtaining detailed medical information about relatives cannot be overemphasized. In one German study, only 20% of patients with some form of familial glomerulonephritis were aware of renal disease in their relatives.” The same study found that AS accounted for 50% of all familial glomerulonephritis.
(2) Renal biopsy Light
Microscopy contributes little towards a diagnosis of AS. The results are normal in children under the age of 10, and even in adult life the findings are non-specific. They may include segmental sclerosis and obsolescence, tubular atrophy, interstitial fibrosis, and infiltration by lymphocytes and plasma cells with clusters of foam cells. An experienced pathologist may recognize thickening of the glomerular capillary walls by light microscopy [6].
(3) Characteristic ophthalmic signs
The ophthalmic manifestations of AS were first reported in 1954, [7] and the characteristic triad of signs is well recognized by ophthalmologists [8] but only visible using a slit lamp ophthalmoscope. Patients with progressive renal disease and eye abnormalities in the absence of hearing problems are very rare [9], as deafness usually precedes any ophthalmic signs.
The three characteristic features are: (1) anterior lenticonus, (2) macular flecks, and (3) peripheral coalescing flecks.
Patients may have one or more of these. Some authors believe that the anterior lenticonus is diagnostic of AS [8,10], as all the patients with anterior lenticonus whom they studied had progressive renal disease. Anterior lenticonus causes slowly progressive axial myopia, and rarely may it progress to anterior capsular cataract, for which surgical extraction is needed. (Posterior lenticonus is not specific to AS.) Occasionally, lens opacities have been described in AS, but they are not specific. White perifoveal flecks are also characteristic of AS. They do not affect vision and fluorescein angiography of the macula is normal.
(4) High-tone sensorineural deafness
The detection of sensorineural deafness in a patient with hematuria should always suggest a diagnosis of AS, even in the absence of a positive family history or the diagnostic eye signs. It is important to perform an audiogram on any patient with unexplained hematuria even if the family history is negative, as the hearing loss may be subclinical at first, although it is usually bilateral.
Deafness is often progressive during childhood, particularly in males [11,12], eventually necessitating the use of a hearing aid [13].
No definite treatment exists for Alport’s syndrome. Supportive treatment for Alport’s syndrome includes ACE inhibitors, which have been used to treat hypertension as well as reduce proteinuria. Cyclosporine has also been used to halt disease progression in those patients with severe proteinuria [14]. In those patients with end-stage renal disease, both dialysis and transplantation are options; however anti-glomerular basement membrane disease can develop in 3-4% of transplanted patients [15]. Gene therapy for Alport’s syndrome is being studied. Animal studies are underway to evaluate the delivery of human alpha-5 (IV) chain of GBM in a canine model of X-linked Alport’s syndrome [16].
Based on typical triad of positive family history, anterior and posterior lenticonus sensorineural deafness and medical renal disease, a diagnosis of Alport’s syndrome was made. The patient was treated with diuretics, antihypertensive agent, iron replacement, and hearing aids.
CONCLUSION
It is important to recognize Alport’s syndrome early in the course of the disease. This is facilitated by an integrated approach to diagnosis. The role of an ophthalmologist is prudent in the diagnosis of such interdisciplinary pathological entities. There should be a high index of suspicion for Alport’s syndrome in any patient presenting with anterior and posterior lenticonus. Early diagnosis can improve longevity and improve the prognosis of Alport’s syndrome patients.
DECLARATIONS
Patient consent: Written informed consent was obtained from the patient for participation publication of this case report and any accompanying images.
Competing interests: The authors declare that they have no competing interests.
Data availability: The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Contributor: RK did the workup of the case. AQ described the ocular changes and wrote the manuscript. TD supervision and review. All authors read and approved the final manuscript.
REFERENCES
1. Flinter F. Alport’s syndrome. J Med Genet. 1997; 34: 326-330.
6. Guthrie LG. “Idiopathic” or congenital hereditary and family haematuria. Lancet. 1902-i: 1243-1246.
9. Grunfeld JP. The clinical spectrum of hereditary nephritis. Kidney Int. 1985; 27: 83-92.
10. Nielsen CE. Anterior lenticonus and Alport’s syndrome. Acta Opththalmol (Copenh). 1978; 56: 518-530.






































































































































