A Pilot Study of a Potential Phosphopantothenate Replacement Therapy in 2 Patients with Pantothenate Kinase-Associated Neurodegeneration
- 1. CEDIMAT, Santo Domingo, Dominican Republic
- 2. Retrophin, Inc., Cambridge, MA, USA
About the Corresponding Author
Dr. Pedro Roa
Summary of background:
Neurology Residency at HSBG - Universidad Autonoma de Santo Domingo. Head of Neurology at CEDIMAT. Professor of Neurology at Universidad Iberoa mericana. Director of PKAN - CEDIMAT Research Project in Dominican Republic. Member of American Academy of Neurology and Dominican AssociaJ on of Neurology.
Website: https://pkanenrd.jimdo.com
Permanent e-mail address: proa@cedimat.net
Current research focus:
• Phosphopantothenate replacement therapy in 2 patients with PKAN.
• Grey matter alterations in patients with PKAN.
• Acanthocytosis and the c.680 A>G Mutation in the PANK2 Gene.
• Missense PANK2 mutation without “Eye of the tiger” sign: MR findings in a large group of patients with PKAN.
• Neurologic Improvement aYer a hypercaloric diet in two patients with PKAN.
ABSTRACT
Pantothenate kinase-associated neurodegeneration (PKAN) is an autosomal recessive disorder linked to mutations in the PANK2 gene, presenting with mixed-motor symptoms of dystonia and parkinsonism in childhood to early adulthood. The pantothenate kinase 2 (PanK2) enzyme phosphorylates pantothenic acid to form phosphopantothenic acid (PPA), a key step in the biosynthesis of coenzyme A (CoA). We describe the treatment of 2 adult male siblings with PKAN under compassionate use authorization with fosmetpantotenate (RE-024), a molecule designed to pass the blood-brain barrier and be metabolized to form PPA in neural and glial cells. The therapy aims to restore PPA, the product of the dysfunctional PanK2 enzyme, to physiologic levels. Open, uncontrolled fosmetpantotenate treatment was associated with clinically meaningful improvement, including improved gait and overall functioning in the first 6 months, followed by stabilization of disease progression through 47 weeks. A controlled trials should be conducted to evaluate the safety and efficacy of fosmetpantotenate in patients with PKAN.
KEYWORDS
• PANK2 gene
• Parkinsonism
• Dystonia
• Fosmetpantotenate
• Pantothenate kinase-associated neurodegeneration
CITATION
Roa P, Stoeter P, Perez-Then E, Santana M, Marshall R (2017) A Pilot Study of a Potential Phosphopantothenate Replacement Therapy in 2 Patients With Pantothenate Kinase-Associated Neurodegeneration. Int J Rare Dis Orphan Drugs 2(2): 1006
ABBREVIATIONS
BAD: Barry-Albright Dystonia; CEDIMAT: Centers for Diagnostic and Advanced Medicine and Medical Conferences and Telemedicine; CoA: Coenzyme A; ECG: Electrocardiogram; F-M: Fahn-Marsden; MDS-UPDRS: Movement Disorder Society-Unified Parkinson's Disease Rating Scale; MRI: Magnetic resonance imaging; PanK2: Pantothenate kinase 2; PKAN: Pantothenate kinase-associated neurodegeneration; PPA: Phosphopantothenic acid
INTRODUCTION
Pantothenate kinase-associated neurodegeneration (PKAN) is an autosomal recessive disorder with a variable clinical phenotype and a prevalence of 1 to 3 per million persons worldwide [1]. Clinical manifestations of mixed-motor symptoms include focal and generalized dystonia, parkinsonism, spasticity, dysarthria/anarthria, and dysphagia, often requiring gastric tube placement [1]. The genotype/phenotype association is not well understood, in that key features such as rate of progression, age of onset, and signs and symptoms are highly variable, even among siblings and case clusters with identical mutations [2]. Current therapeutic strategies for PKAN rely on symptomatic relief [1].
The normal product of the PKAN causative gene, PANK2, is the enzyme pantothenate kinase 2 (PanK2). PanK2 phosphorylates pantothenic acid to form phosphopantothenic acid (PPA), a key step in the biosynthesis of coenzyme A (CoA) [3]. It has been postulated that the defective PanK2 enzyme, localized primarily in mitochondria, has decreased ability to phosphorylate pantothenate, thus leading to decreased concentrations of CoA in selectively vulnerable tissues, including brain tissue [4]. CoA is essential to biochemical reactions that impact energy metabolism, membrane integrity, signaling, and other critical processes [3].
Fosmetpantotenate (RE-024) is a molecule that is designed to be metabolized to PPA intracellularly, including to neural and glial cells, aiming to restore PPA and therefore CoA levels to address the molecular defect directly in patients with PKAN. The mechanism of action and potential to reach the brain in mammalian species have been studied. Fosmetpantotenate has been shown to significantly increase intracellular CoA levels as a primary marker of its activity and to increase tubulin acetylation as a marker of the action of CoA in vitro in neuroblastoma cells that have been silenced for PanK2 expression [5,6]. Additionally, PPA generated by orally dosed fosmetpantotenate in mice reaches the brain and is incorporated into CoA [5,6]. When dosed orally in nonhuman primates and sampled using microdialysis techniques, fosmetpantotenate was found in the brain at levels consistent with meaningful conversion to PPA [6].
Thus, a clinical pilot study appeared to be justified. Here, we describe the treatment and response of 2 adult male siblings with PKAN, who tested homozygous for the c.680 A>G mutation in the PANK2 gene. This mutation replaces a tyrosine by a cysteine at position 227 in PanK2 [7]. Their advanced disease was treated with fosmetpantotenate under compassionate drug use approval by the National Council of Bioethics in the Dominican Republic and the Centers for Diagnostic and Advanced Medicine and Medical Conferences and Telemedicine (CEDIMAT) Institutional Review Board.
CASE PRESENTATION
MEDICAL HISTORY
Patient 1
Course of Illness: Symptoms presented at age 10 years, with frequent falls while walking and running at school. The patient developed cervical dystonia and abnormal movements, and posturing of the hands and feet, leading to great difficulty in writing and walking. Symptoms progressed to dysphagia, oropharyngeal dystonia with tongue protrusion, increasing difficulty articulating words, and drooling. From age 12, he developed increasing difficulty with activities of daily living, such as dressing and maintaining personal hygiene. School teachers reported cognitive decline and abnormal behavior characterized by extreme “out of control” impulsivity. He required support for ambulation from age 18, and by age 23, choking while eating was a medical concern. PKAN was diagnosed at age 20 by clinical presentation, MRI (“eye-of-the-tiger sign”), and genetic testing.
Previous Treatment: Previous treatments included botulinum toxin injections in the calves at age 21 to reduce dystonia; the injections were temporarily beneficial for approximately 3 months. Concomitant medications were risperidone 0.3 mg daily, with some benefit for impulsivity.
Baseline Presentation Prior to Treatment: At age 24, manifestations of PKAN included orofacial and tongue dystonia with associated drooling, severe foot dystonia, great difficulty walking except with support, moderate to severe upper-limb dystonia, marked bradykinesia, dysphagia, and severe dysarthria. There were no other complaints, and his body mass index was within normal limits over the whole study (around 20 kg/m²).
Patient 2
Course of Illness: Symptoms date from age 10 years, when the patient developed frequent falls, hand dystonia, bradykinesia, and orofacial dystonia. Orobuccolingual dystonia with tongue protrusion resulted in drooling and increasing difficulty eating and articulating words by age 18. Hand movements became increasingly clumsy due to dystonia, and writing deteriorated. He became unable to speak or walk, and required assistance for most activities of daily living by age 23. A gastric tube for feeding was placed at age 26. Management of the patient was increasingly difficult due to extreme impulsivity, poor tolerance of frustration, and occasional pushing and hitting of his brother or parents. PKAN was diagnosed by clinical presentation, magnetic resonance imaging (MRI) (“eye-of-the-tiger sign”), and genetic testing at age 24.
Previous Treatment: Previous treatments included botulinum toxin A injections in the calves at age 24 to reduce dystonia; the injections were temporarily beneficial for approximately 3 months. Concomitant medications were risperidone 2.5 mg daily; clonazepam 0.5 mg twice daily; trihexyphenidyl 2.5 mg twice daily; mefenamic acid 500 mg as needed; and propinox 10 drops as needed.
Baseline Presentation Prior to Treatment: At age 29, manifestations of PKAN included orofacial and tongue dystonia with constant drooling, severe foot dystonia, inability to walk except with strong support, moderate to severe upper-limb and trunk dystonia, marked bradykinesia, freezing, dysphagia, and severe dysarthria. His body mass index stayed low during the study, changing between 13.5 and 14 kg/m².
TREATMENT
After approval from local and national authorities, treatment with oral fosmetpantotenate was initiated. Treatment results through 47 weeks are described.
Fosmetpantotenate Treatment of Patients 1 and 2: An initial target dose of 1 mg/kg given 3 times daily was selected based on animal safety data and on the known half-life of phosphopantothenate (~4 hours). This dose represented a 175- fold margin of safety with respect to the NOAEL (no adverse effect limit) in rats. Thus, fosmetpantotenate was increased in Patient 1 from 60 mg (1 mg/kg in a 60-kg patient) to 180 mg daily (60 mg 3 times daily), and in Patient 2 from 40 mg (1 mg/kg in a 40- kg patient) to 120 mg daily (40 mg 3 times daily) over a 5-day period, followed by daily dosing at 180 mg (60 mg 3 times daily) and 120 mg (40 mg 3 times daily), respectively. Baseline severity and treatment response were assessed by a neurologist, based on the Movement Disorder Society-Unified Parkinson's Disease Rating Scale (MDS-UPDRS) [8], the Barry-Albright Dystonia (BAD) Scale [9], and the Fahn-Marsden (F-M) Scale [10]. Videos of the patients walking were also captured periodically. Routine chemistry, hematology, and coagulation laboratory assessments as well as electrocardiograms (ECGs) were conducted at each clinic visit to monitor safety
Video 1: Video illustrates the baseline gait and improvement in walking after 12 and 40 months of treatment with fosmetpantotenate in 2 adult male siblings with Pantothenate kinase-associated neurodegeneration (PKAN).
RESULTS
Safety monitoring through repeated laboratory assessments (chemistry, hematology, and urine analysis) and ECG were unremarkable for obvious treatment-related adverse events. Both patients had a mild anemia and low eosinophilic cell counts at baseline and throughout the study. Thrombin time was slightly elevated repeatedly in Patient 1 (16 to 18 seconds) and occasionally some instances in Patient 2. There was a slight increase in creatine kinase (170–200 units), mainly in Patient 1, who also showed an increase in serum lactate up to 29 mg/ dL and slightly elevated directly measured bilirubin levels up to 0.29 mg/dL on several occasions. Cholesterol, however, was normal. Patient 2 showed low levels of serum creatinine (down to 0.47 mg/dL) for some months, which normalized during the end of the study. In summary, none of these slight deviations in laboratory values were considered adverse events, and no other treatment-related adverse events were observed during the 47- week period. MRI examinations done before treatment and after approximately one year showed a slight drop in the T2-Time in the globus pallidus from 32.7±4.9 ms to 31.8±4.0 ms in Patient 1 and from 30.4±5.7 ms to 28.5±2.6 ms in Patient 2.
Clinical Response Patient 1
The patient experienced gradual improvement in his gait, walking consistently unassisted for short distances within 8 weeks of treatment. Gait continued to improve gradually over several months of treatment. The family and physician reported that impulsivity dramatically improved, allowing discontinuation of risperidone. The orofacial dystonia showed improvement within the first week and remained improved over 47 weeks, with more control over tongue protrusion, less open jaw dystonia, and somewhat better articulated speech. Hand dystonia and writing improved as well. Apart from 2 episodes of obvious distress with reduced ability to walk without assistance due to transient infections (one was accompanied by coughing and the other by diarrhea), the clinical course was stable. At 47 weeks, standing, walking, and postural stability remained improved, and more spontaneous speech was reported by the family. Observed clinical improvement from baseline to 47 weeks of treatment was also reflected in improvement on the MDS-UPDRS Parts II (27, 16; 41%) and III (56, 44; 31%) (Figures 1a and 1b). More spontaneous speech has been noted by the family.
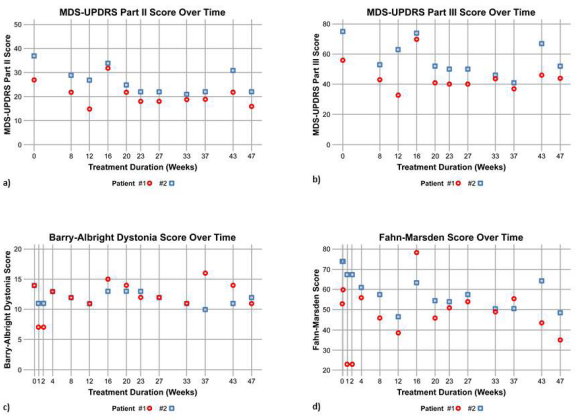
Figure 1: a) MDS-UPDRS Part II and b) Part III; response observed on the c) BAD Scale and d) F-M Scale.
Clinical Response Patient 2
The patient experienced gradual improvement in his gait, showing ability to walk with assistance and then without assistance for short distances within 8 weeks of treatment. His gait continued to improve over several months of treatment. The family reported that impulsive behavior and frustration tolerance were also notably improved. There was one episode of distress, mainly of deterioration of free walking, without relation to an accompanying infection or secondary disease. During the last weeks of the study, the patient was more attentive and interactive, and more independent and able to get out of bed unassisted. Observed clinical improvement from baseline to 47 weeks of treatment was also reflected in improvement on the MDS-UPDRS Parts II (37, 22; 41%) and III (75, 52; 21%) (Figures 1a and 1b). A video of both patients walking before and after treatment is also included. Both patients have elected to continue treatment with fosmetpantotenate due to perceived benefit.
DISCUSSION
The natural history of these 2 sibling patients showed common findings of onset in late childhood, parkinsonism, dystonia, decreased quality of life, and greatly impaired functioning with activities of daily living. Open, uncontrolled fosmetpantotenate treatment over 47 weeks was associated with clinically meaningful improvement of axial and limb symptoms overall, superimposed on the variable natural course of the disease, followed by stabilization of disease progression. Improvement in gait was especially notable and consistent with improvement on the MDS-UPDRS Part II, which correlates well with other disability and quality-of-life measures [11], and on Part III, which is highly correlated with Part II [8]. Mean improvement across both patients at 23 weeks was 38% on Part II and 31% on Part III. These scales assess functional impairment in activities of daily living (II) and neurologic impairment (III) typically seen in movement disorder patients across disease states, and therefore have broader applicability than originally conceived.
The response observed on the BAD and F-M scales (Figures 1c and 1d) was less obvious and inconsistent with the overall clinically observed course. The BAD and F-M scales have significant limitations, most notably the smallest detectable differences of 17.72% and 27.39%, respectively, suggesting that the properties of both scales may limit their utility for detecting the full range of clinical improvement in PKAN clinical trials [12].
No clinically significant laboratory abnormalities were noted. The slight elevation of lactate seen in Patient 1 may be explained by mitochondrial dysfunction, and impaired bile acid metabolism has been described as well in PKAN [13]. The slight elevation of creatinine kinase might be due to increased muscular efforts to compensate for dystonic movements, but without obvious correlation to the distress periods. The mild anemia mainly seen in Patient 2 was attributed mainly to his reduced nutritional state, because his level of acanthocytes measured previously was normal [7].
Current standard of care in PKAN is focused on symptomatic relief, and is based on small open trials, rational therapy of dystonia and parkinsonism, and clinical experience rather than adequate and well-controlled studies [14]. Deferiprone, an iron chelating agent currently under study in an international control trial, was reported to produce reduction in iron accumulation in the pallidum [15], and induced clinical stabilization in 5 out of 6 patients over 4 years [16]. However, the association between brain iron accumulation and phenotype is not well understood, and may not be a reliable indicator of treatment effect, at least for fosmetpantotenate [17].
Case series and reports have also reported partial improvement with deep brain stimulation [18,19], transcranial magnetic stimulation,[20] and botulinum toxin [21]. Preclinical research has generated additional disease-modifying strategies regarding replenishing CoA via various entry points in the metabolic pathway, including supplementation with pantethine [22] or with CoA itself [23]. Orellana et al found that mitochondrial and neuronal functionality could be restored by CoA supplementation in cultured neurons derived from stem cells from PKAN [23].
Finally, Christou et al [24] recently reported a 12-month, open-label treatment with fosmetpantotenate of an adult male with later-onset PKAN [24]. The authors observed continuous improvement in the patient’s gait over the first 2 months, followed by stabilization, and also noted improvements in the UPDRS and BAD scales, as well as in quality of life. This patient experienced an initial elevation in liver function tests, which resolved after discontinuation followed by a reduced dose strategy, while maintaining the observed gains in motor function. Overall, findings are similar to our 2 patients, although our patients appear to demonstrate more variability of symptoms over the 1-year period.
CONCLUSION
Although the natural history of the disease has not been well documented in longitudinal studies, and patients with PKAN do experience fluctuations as well as periods of stable disease, this degree of improvement followed by stabilization is not typical of the natural history of the disease in either the early- or later-onset phenotypes. This judgment is based, in part, on our extensive experience with more than 40 patients with PKAN who we follow in the Dominican Republic. The pattern of response suggests that most or all of the clinical improvement associated with fosmetpantotenate treatment may be seen over approximately a 6-month period, with subsequent stabilization of disease progression. Our findings are similar to those of an independent research group [24]. However, placebo response, or an unusual natural history course in these 2 patients, cannot be ruled out in an uncontrolled setting for both of these case reports. Controlled trials are therefore needed to evaluate the safety and efficacy of fosmetpantotenate in patients with PKAN. An international, randomized, double-blind, placebo-controlled trial of fosmetpantotenate is planned to initiate in 2017 [25].
ACKNOWLEDGEMENT
We would like to thank the patients and family who participated, and in particular Amanda Harring-Abbott, Kristyn Bogli and Katherine Lucey for their efforts in conducting this work. We are also grateful to the anonymous reviewers for their comments and peer-review
DISCLOSURE
CEDIMAT was the recipient of an unrestricted grant from Retrophin, Inc. to support research and treatment in PKAN. Drs. Roa, Perez-Then, and Santana declare no financial disclosures. This study was presented as a poster presentation at the International Congress of Parkinson’s Disease and Movement Disorders Annual Meeting, June 19-23, 2016, Berlin, Germany. Editorial support for this manuscript was provided by Scientific Communications Group and was sponsored by Retrophin, Inc.








































































































































