Laron Syndrome
- 1. Endocrinology and Diabetes Research Unit, Schneider Children’s Medical Center, Tel Aviv University, Israel
Abstract
Laron syndrome (LS) or primary growth hormone (GH) insensitivity is caused by deletions or mutations in the GH-receptor gene. Its clinical characteristic is dwarfism, acromicria, obesity and protruding forehead. Patients homozygous for these gene defects are protected from cancer lifelong.
Keywords
• Laron Syndrome
• GH Receptor Defect
• Primary GH Insensitivity
• Congenital IGF-I Deficiency
Citation
Laron Z, Kauli R (2022) Laron Syndrome. Int J Rare Dis Orph Drugs 5(1): 1011.
ABBREVIATIONS
GH: Growth Hormone, GH-R: Growth Hormone Receptor, IGF-I: Insulin Like Growth Factor
INTRODUCTION
Laron syndrome (LS) or primary growth hormone insensitivity (OMIM# 262500) was first reported in 1966 and 1968 in very short children products of newly immigrated Jews from Yemen [1,2] (Figure 1).
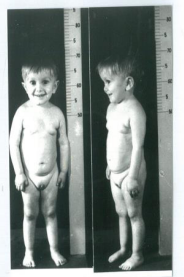
Figure 1: Typical features of 3½ years old boy with Laron Syndrome.
All belonged of consanguineous families and resembled congenital GH deficiency. However with the introduction of the GH radioimmunoassay [3] it was found that the levels of their serum GH were very high. Furthermore administration of exogenous GH did not cause an elevation of the serum IGF-I levels, demonstrating GH insensitivity [4].
Following our publications, many patients with LS were reported, from other countries and continents. Genetic analysis of our patients [5,6] and those from others revealed a series of different mutations and deletions in the GH-(receptor) gene, varying in different geographical areas [7,8]. The finding of the same mutations (E180) in a Moroccan Jewish patient, as well as the patients from South America [8] raised the hypothesis that all belonged to Jews fleeing the Spanish inquisition [9]. The finding of an 18000 year old female skeleton on the Island of Flores resembling the bone X-rays of LS patients led to the assumption that the founder gene for LS was in Indonesia [10].
MATERIALS AND METHODS
Review of medical records from our clinic and literature.
RESULTS AND DISCUSSION
Since our first descriptions, we have followed 76 LS patients, many from early childhood into adult age. The total number of LS patients worldwide is estimated to be around 500.
|
Table 1: Main clinical and laboratory characteristics. |
|
|
Clinical |
Laboratory |
|
Dwarfism (height -4-10 SDS) |
Hypoglycemia in infancy |
|
Obesity |
High serum hGH |
|
Spare hair |
Low to undetectable IGF-I |
|
Small head circumference |
Low IGFBP-3 |
|
Frontal bossing, sunset sign |
Serum GHBP (- or +) |
|
Crowded, defect teeth |
Progressive hyperlipidemia |
|
Acromicria (small chin, hands, feet) |
|
|
Small gonads and genitalia |
|
|
High pitched voice |
|
|
Retarded skeletal maturation |
|
|
Slow mot or development |
|
The clinical and laboratory date of LS patients have been described in detail [11,12]. The main clinical and laboratory characteristics are shown in Table 1. Already at birth LS infants are obese, and the degree of obesity increases to extremes in adult age when 59±5% of the body composition in females and 39±6% in males are adipose tissue [13] (Figure 2).
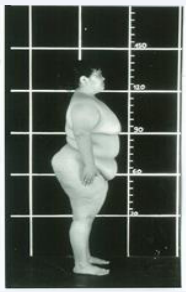
Figure 2: Obesity of a 19 year old female patient with Laron Syndrome.
The linear growth deficit ranged between -4 to -10 SDS height. Using the growth pattern of untreated patients, special growth charts for LS were, designed [14].
Imaging of the skeleton show under development of the facial bones and bone age retardation. Skull CT and MRI have revealed a series of abnormalities such as absence of sinuses, spinal stenosis and in some patient’s variable diffuse parenchymal loss of the brain [15]. These abnormalities affected the academic performance in those patients [16].
Due to the high degree of obesity, LS patients develop glucose intolerance and some even diabetes with its complications.
Treatment
The only available treatment is daily subcutaneous injections of IGF-I which stimulates growth but to a lesser degree than GH in GH deficient patients [17]. Is also decreases serum GH and insulin [18].
Adverse Effects (AE)
Acute effects are hypoglycemia are water retention and intracranial hypertension. Long-term treatment causes further obesity and hyperandrogenism [19]
All adverse effects are reversible with reduction of the dose of IGF-I or stopping treatment.
Cancer
One unexpected observation was that LS patients homozygous for the GH-R defects are protected lifelong from development of cancer [20] heterozygous relatives are not [21].
Ongoing genetic investigations using immortalized lymphoblastoid cells from LS patients revealed that LS patients expressed high levels of tumor suppressor genes and low levels of oncogenic proteins [22].
CONCLUSION
Laron Syndrome is a unique model to study the biologic and metabolic effects along GH/GF-I axis and the differential actions between Growth Hormone and IGF-I.
ACKNOWLEDGEMENTS
The author wishes to acknowledge Prof. Rivka Kaul, Dr. Beatrice Klinger and Prof. Haim Werner for collaboration throughout many years of study.
REFERENCES
6. Shevah O, Laron Z, Genetic Aspects in Laron Syndrome – Man to Mouse Laron Z, Kopchick J (eds). Springer-Verlag Berlin Heidelberg 2011; 29-52.
11. Z Laron, J Kopchick (Eds). Laron Syndrome – From Man to Mouse. Springer-Verlag Berlin Heidelberg 2011; 513.
16. Laron Z. Adjustment and rehabilitation problems of children, adolescents, and adults with Laron syndrome in Laron Syndrome – From Man to Mouse, Laron Z, Kopchick J (Eds). Springer-Verlag Berlin Heidelberg. 2011; 335-337.








































































































































