Neonatal leukemia cutis presenting with dysmorphic features and cutis laxa
- 1. Department of Maternal and Child Health, Division of Neonatology and Neonatal Intensive Care Unit, Santa Chiara University Hospital of Pisa, Italy
- 2. Department of Clinical Medicine, Santa Chiara University Hospital of Pisa, Italy
- 3. Department of Maternal and Child Health, Division of Pediatric Onco-Hematology, Santa Chiara University Hospital of Pisa, Italy
- 4. Department of Medicine of Laboratory, Laboratory of Cytogenetic, Santa Chiara University Hospital of Pisa, Italy
About the Corresponding Author
Dr. Francesca Dini
Summary of background:
Pediatrician, postgraduated at University of Pisa, Italy. I have worked for 2 years in the Neonatal Intensive Care Unit of Pisa's Hospital, Italy. Currently, I work in the Pediatric Unit of Lucca's Hospital, Italy. I deal especially of neonatal endocrinological diseases.
Permanent e-mail address: francesca.dini@hotmail.com
Current research focus:
• Evaluation of prevalence and pre and post natal factors related to premature neonates, EUGR, SGA; their follow up.
• Evaluation of the tyroid status in premature neonates; evaluation of sensibility of ‘TSH screening’; definition of Ioduria in the newborn.
ABSTRACT
Congenital leukemia is a rare disease with particular biological and clinical characteristics, which differs from those of older children. Its prognosis is generally poor. Its clinical manifestation may vary (hyperleukocytosis, thrombocytopenia, organomegaly) and some patients can develop cutaneous infiltration by leukemic cells (leukemia cutis). We describe a dysmorphic patient with thrombocytopenia hiding a congenital leukemia with fatal outcome. At birth he presented cutis laxa, multiple dysmorphic, thrombocytopenia and hepatosplenomegaly, initially orienting towards the diagnosis of a syndrome. Afterwards, pancytopenia and coagulopathy led to the diagnosis of congenital leukemia. His clinical features didn’t fit with any of the syndromes described in literature as associated with an increased risk of leukemia (i.e. Down syndrome, Fanconi’s anemia). This suggests a possible new association between a severe neonatal leukemia cutis and a dysmorphic syndrome characterized by cutis laxa (i.e. TALDO deficiency?).
KEYWORDS
• Leukemia
• Cutis laxa
• Newborn
• Thrombocytopenia
• Dysmorphic features
CITATION
Dini F, Tuoni C, Vannozzi I, Toschi B, Alberti E, Nardi M, Bertini V, Valetto A, Giampietri M, Vuerich M, Ciantelli M, Boldrini A, Ghirri P (2017) Neonatal leukemia cutis presenting with dysmorphic features and cutis laxa. Int J Rare Dis Orphan Drugs 2(1): 1005
INTRODUCTION
Leukemia is the most frequent malignancy in childhood. The majority of cases are acute myeloid leukemia, while lymphoblastic leukemia most often is on B cell lineage [1]. Leukemia may cause significant coagulopathy, putting the patient at risk of intracranial haemorrhage [2].
Congenital leukemia is a rare disease (<1% of all leukemia in childhood). This particular type of leukemia has peculiar findings that differ from those of older children. The clinical presentation may vary. The newborns may present the classic findings of hyperleukocytosis associated with hepato-splenomegaly, or can present purplish nodules as initial manifestation (leukemia cutis). Approximately 10% of patients present cutaneous leukemia without bone marrow involvement (aleukemic leukemia cutis) [3,4].
The majority of congenital leukemia are associated with cytogenetic disorders that often involve rearrangements of the mixed lineage leukemia (MLL) gene (11q23): gene translocations give rise to fusion proteins that increase the expression of genes encoded for transcription factors involved in haematopoiesis. The presence of MLL translocations leads to aggressive acute leukemia with extremely poor prognosis [3,5]. An association has been described between leukemia and some inherited conditions such as Down syndrome, neurofibromatosis, Bloom’s syndrome, Fanconi’s anemia [6] and Wiskott-Aldrich syndrome [7].
CASE PRESENTATION
We report the case of a full-term male newborn delivered from a mother affected by unspecified thrombocytopenia. The antenatal period was uneventful, except for intra-uterine growth restriction (IUGR). He was the first born of healthy parents, with uncertain consanguinity. The five-minute Apgar score was 8 and birth weight was 2450 g (<10°centile) [8]. The baby was noted at birth to have spread petechiae and ecchymosis. He displayed dysmorphic features in the form of hypertelorism, depressed nasal bridge, prominent upper lip, small chin, shawl scrotum, varus right foot, excessive skin folds, especially at base of the limbs, multiple infiltrated plaques on the scalp, sparse thin hair. Laboratory test showed coagulopathy (fibrinogen 87.4 mg/dl, undetectable values for prothrombin time and partial thromboplastin time) and severe thrombocytopenia (PTL 8,000/ mcl) with haemoglobin (Hb) level and white blood cells (WBC) count within normal ranges. Viral serology for EBV, HSV, CMV and toxoplasmosis was negative. ABO or Rh incompatibility was excluded. Hepato-splenomegaly was observed clinically and on the abdominal ultrasound scan; cranial ultrasound scan, eye examination and earing tests resulted normal.
From day 5, he started to develop anaemia (RBC 2,690,000/ mcl, Hb 8.9 gr/dl, Hct 24.6%). A peripheral blood smear was also performed, showing normal features.
During the hospital stay, the baby received 8 PTL concentrate transfusions (HLA-compatible), 2 packed RBC transfusions and 1 fresh frozen plasma transfusion; prednisone (4 mg/kg/day for 3 days) and intravenous immunoglobulin (IVIG) (1 gr/kg/day for 3 days) were also administered.
Thus, we performed genetic testing to investigate on resistant thrombocytopenia in a syndromic setting. In our newborn, the presence of cutis laxa, dysmorphic features, hepatosplenomegaly, anaemia and thrombocytopenia suggested a possible diagnosis of transaldolase deficiency, a recently described inborn error of pentose phosphate pathway.
Afterwards, his clinical condition rapidly worsens with jaundice, hypocolic stools and hyperchromic urine (direct bilirubin 7.05 mg/dl), oral mycosis, Campylobacter enteritis, Klebsiella pn sepsis. At 17 days of life multiple purple nodules were scattered over his chest, arms and legs and a biopsy was performed (Figure 1).
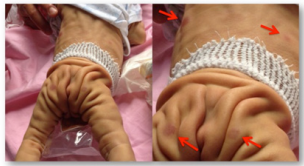
Figure 1: Clinical features of our patient showing extensive cutis laxa (present from birth), multiple purple nodules predominantly on the trunk (appeared on day 17 of life).
In few days his haematochemical profile deteriorated (RBC 1,340,000/mcl, Hb 4.0 gr/dl, PTL 5,000/mcl, WBC 4,630/mcl with blasts 45%, potassium 8.49 mEq/L, AST 303 UI/L, ALT 42 UI/L, GGT 70 UI/L, LDH 2.511 UI/L, prothrombin time 17%, aPTT 65.9 sec, fibrinogen 186 mg/dl, antithrombin III 18%, D-dimer test 10.53 mcg/ml). In the suspect of a leukemic disease a bone marrow examination was performed, confirming the diagnosis. Rapidly, he became drowsy, hypothermic, hypotensive with bradycardia and successor apneas, not responsive to resuscitation. The patient died before chemotherapy was started.
The results of the skin biopsy were available post-mortem showing a pattern positive for CD45/LCA, PAX-5, CD99, TdT-/+, CD79a+/-. CD10 was negative. This setting is compatible with lymphoblastic leukemia with ambiguous immunophenotype, broadly classified into B-lineage. Karyotype and 8x60k array CGH (Agilent, Santa Chiara, CA) have been performed at birth to detect genetic imbalances for the presence of syndromic features with cutis laxa. They detected multiple imbalances (Table 1), especially on chromosome 7 and 11, probably due to blast cells.
Table 1: Genetic imbalances detected by array CGH are shown; the chromosome (chr), the cytoband, the relative position (hg19 build) and the imbalance extent (kb) are given.
| Aberration | Chr | Cytoband | Start | Stop | Extent (kb) | Type |
| 1 | chr7 | p22.1 - p12.1 | 5,617,816 | 51,182,300 | 120 | loss |
| 2 | chr7 | q11.23 - q22.1 | 72,726,578 | 98,927,336 | 45,564 | loss |
| 3 | chr7 | q36.1 - q36.3 | 148,774,870 | 158,909,738 | 26,201 | loss |
| 4 | chr9 | p13.2 - p13.1 | 36,749,801 | 39,156,954 | 10,134 | loss |
| 5 | chr9 | q33.3 - q34.11 | 129,571,350 | 130,825,596 | 2,407 | loss |
| 6 | chr11 | q14.1 | 84,245,609 | 84,367,297 | 121 | loss |
| 7 | chr11 | q23.3 - q25 | 118,339,458 | 134,868,407 | 16,528 | loss |
| 8 | chrX | q13.3 | 74,494,014 | 74,649,810 | 155 | gain |
DISCUSSION
Neonatal leukemia is a rare condition (1-8.6/106 live births) occurring in the first 4 weeks of life. Since its incidence is low, clinical and biological characteristics results difficult to define. Leukemia cutis, a proliferation of leukemic cells in the skin, occurs in 25-64% of patients with neonatal acute leukemia [4]. The most typical clinical features of neonatal leukemia are hepatosplenomegaly and skin lesions as nodular cutaneous infiltrates (leukemia cutis), macules, papules, nodules, haemorrhagic plaques, erythema, or generalized eruption. These lesions frequently present a greenish or bluish colour, giving an appearance of “blueberry muffin baby” [3]. This clinical presentation can be observed from the beginning of the disease or develop latterly, delaying the diagnosis. The presence of skin lesions, associated with pathological haematological profile, guides the diagnosis, which can be confirmed by skin biopsy.
A differential diagnosis of these cutaneous lesions includes intrauterine infections comprising the TORCH syndrome (Toxoplasmosis, Rubella, Cytomegalovirus, Herpes simplex, Coxsackie virus, Parvovirus, Epstein Barr virus, Syphilis) [9], haemolytic disease of the newborn (ABO or Rh incompatibility), neonatal erythematous lupus, and certain neoplasia such as metastatic neuroblastoma or Langerhans cell histiocytosis [4].
In our case, the presence of maternal thrombocytopenia and the suspect of a syndromic pattern confused the diagnosis. Firstly, we excluded a severe form of maternal autoimmune thrombocytopenia, evaluating the absence of anti-PTL antibodies with the failure of steroid and IVIG therapy
Transaldolase deficiency is a heterogeneous disorder of carbohydrate metabolism characterized clinically by dysmorphic features, cutis laxa, hepatosplenomegaly, hepatic fibrosis, pancytopenia, renal and cardiac abnormalities.
The diagnosis can be done by biochemical studies of excretion of polyols in urine, transaldolase enzyme activity on fibroblasts and molecular genetic testing of the transaldolase gene (TALDO1). Transaldolase deficiency is caused by balletic mutations in the TALDO1 gene. In literature 12 cases describing the association between cutis laxa and pancytopenia are reported and they all are ascribable to transaldolase deficiency (Table 2) [10]. Unfortunately, it was not possible to carry out the biochemical analysis of urine for the premature death of the baby and the evaluation of fibroblasts for the skin infiltration of leukemic cells. However, we can look for mutations on his stored DNA.
Table 2: Clinical and molecular features of the TALDO-deficient patients described in literature in comparison to our patient
| Clinical/molecular features of TALDO-deficient patients | Frequency from literature | Our patient |
| (Eyaid W et al. Transaldolase deficiency report of 12 new cases and further delineation of the phenotype) | ||
| Consanguinity | 10/11 | Uncertain |
| Hepatosplenomegaly | 10/11 | Present |
| Anemia | 9/11 | Present |
| Thrombocytopenia | 9/11 | Present |
| Dysmorphism | 8/11 | Present |
| Wrinkly skin | 4/11 | Present |
| Cardiac defects | 8/11 | Absent |
| Neonatal edema | 6/11 | Absent |
| Renal abnormalities | 5/11 | Absent |
| Respiratory dysfunction | 2/11 | Absent |
| Developmental delay | 2/11 | Unknown |
| Abnormal platelets aggregation | 0/11 | Absent |
| Premature death | 5/11 | Present |
CONCLUSION
In our case, the association between cutis laxa and dysmorphic features with a severe neonatal leukemia cutis with bone marrow involvement could be considered a new association between neonatal leukemia and a dysmorphic syndrome (transaldolase deficiency?). Neonatal leukemia is a severe disorder and may present insidiously, delaying the diagnosis. Its evolution can be rapid and aggressive, leading to a therapeutic failure.
ACKNOWLEDGEMENT
We are very grateful to two anonymous reviewers for their comments and peer-review.
DISCLOSURE
The authors declare no conflicts of interest.








































































































































