The First Rare Case of Muscular Atrophy in Libya (Chopra Amiel Gordon Syndrome
- 1. Department of Internal Medicine, Sabha University, Libya
- 2. Department of Pediatrics, Sabha University, Libya
- 3. Department of Pediatrics, Sabha University, Libya
- 4. Department of Internal Medicine, Sabha University, Libya
- 5. Department of Internal Medicine, Traghan Teaching Hospital, Libya
- 6. Department of Pediatrics, Sabha University, Libya
Abstract
Muscular atrophies are a clinically and heterogeneous group of disorders that all share clinical characteristics of progressive muscular weakness and wasted that involve any type of muscles .the cause of muscle atrophy could be due to acquired or genetic (inherited) neuromuscular diseases such as spinal muscular atrophy (called motor neurons) that control muscle movement. Genetic muscle atrophy may affect local region like face such as Chopra-Amiel-Gordon syndrome (CAGS) is were recently implicated in a newly-identified rare intellectual disability syndrome. It is an autosomal dominant disorder characterized by developmental delay and/or impaired intellectual development Speech delay, facial atrophy, and variable other features, Chopra et al., [1]. Here, we present a case of CAGS in a 8-year-old male with remarkable clinical and mental manifestations.
Keywords
Dysmorphic features, Facial atrophy, Heterozygous, Intellectual disability
Citation
Aaedah AA, Jamil AL, Fatima O (2023) The First Rare Case of Muscular Atrophy in Libya (Chopra Amiel Gordon Syndrome). Int J Rare Dis Orph Drugs 6(1): 1012.
INTRODUCTION
Chopra-Amiel-Gordon syndrome (CAGS) is an autosomal dominant disorder characterized by developmental delay and/or impaired intellectual development, speech delay, facial dysmorphism, and variable other features, including recurrent bacterial infections, ophthalmologic abnormalities, and nonspecific brain abnormalities Chopra et al. [1], reported clinical features of 34 patients from 32 families. The patients ranged in age from 4 months to 34 years. The primary features in 31 patients were global developmental delay and/or impaired intellectual development. Here, we present a first rare case of CAGS in Libya for 8-year-old child.
ABBREVIATIONS
CAGS: Chopra-Amiel-Gordon Syndrome; CK: Creatine Kinase; WES: Whole Exome Sequencing
CASE PRESENTATION
Eight years old Libyan boy, from south city of Libya named sabha, complains from gradual progressive right side facial atrophy started at age of two years, begin first at periorbital region then cheek [Figure 1],
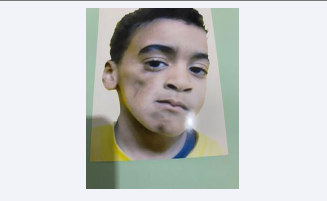
Figure 1: Distribution of pathogenic ANKRD17 variants and 4q13.3 deletions (A) Variants affecting coding sequence.
and mandible ,mother noted that the child had delay in speech confirmed by pediatrician, hearing examination was normal, but he had history of recurrent bacterial infection such as internal otitis media and pneumonia, during pregnancy the mother did not have any chronic diseases and did not have any complications during pregnancy. His intelligence quotient was claimed to be in the normal range. Patient’s family history revealed that one of his brother had deafness, His parents are second degree relatives.
On general physical examination, the child was underweight and short in stature. Standing and walking are normal. Neurological examination of the four limbs is normal and there is no obvious atrophy, The child has stuttering and stammering in words and sentences. Examination of the head is normal, but the face asymmetrical, atrophy of the right side of the face, especially in the cheek and chin area, with hyperpigmentation skin color in those two areas .general dysmorphic features included a triangular-shaped, a high anterior hairline, deep-set eyes, almond-shaped eyes periorbital fullness, thick nasal alae and flared nostrils, The mouth looks normal, but the tongue is small and deviated slightly to the right side.
The patient was subjected to radiological and laboratory investigations. Radiography revealed no grossly abnormalities, non-contrast computed tomography was normal apart of mild atrophy of masticular muscle, decrease volume of parotid gland at right side, the electromyographic examination and muscle biopsy not done because not available. Serological analysis [2] showed creatine kinase (CK) level was normal 78.9U/L, molecular genetic analysis for whole exome sequencing (WES) showing the heterozygous variant c.1309G>A, p.(ALa437Thr),chr4:74013069 in the ANKRD17 gene (OMIM:*615929). This variant leads to an amino acid exchange. pathogenic variants in the ANKRD17 gene (OMIM: *615929) are causative for autosomal dominant Chopra Amiel–Gordon syndrome [Figure 2].
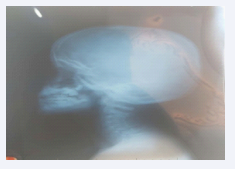
Figure 2: Skull and cervical x-ray of the patient.
The child was advised to consult a pediatrician regarding his general and physical health status. He was counseled to undergo daily physiotherapy, regular assessment for progressive muscle weakness. The patient was kept under periodic recall to prevent any further complications.
DISCUSSION
Chopra-Amiel-Gordon syndrome (CAGS) is an autosomal dominant disorder characterized by developmental delay and/ or impaired intellectual development, speech delay, facial dysmorphism, and variable other features, including recurrent bacterial infections, ophthalmologic abnormalities, and nonspecific brain abnormalities [1].
Chopra et al. [1], reported clinical features of 34 patients from 32 families ascertained using GeneMatcher and DECIPHER. The patients ranged in age from 4 months to 34 years. The primary features in 31 patients were global developmental delay and/or impaired intellectual development. The severity was variable, with 19 patients in the moderate to severe range and 12 in the mild or borderline range. Speech development was delayed in 29 individuals, including 6 with absent speech. Less commonly reported developmental features included autism spectrum disorder and attention deficit- hyperactivity disorder (ADHD). Postnatal microcephaly was reported in 7 patients, and macrocephaly in 4. Epilepsy was reported in 9 patients. Neuroimaging, performed in 23 individuals, identified abnormalities in 11 patients; abnormalities included decreased white matter volume in 3 patients, thinning of the corpus callosum in 2, optic nerve hypoplasia in 2, localized hyperintensity in 2, right temporal sclerosis in 1, dilated Virchow-Robin spaces in 1, and periventricular nodular heterotopia in 1. Ophthalmologic abnormalities were reported in 13 of 23 individuals [3]. Nine patients had recurrent bacterial infections, 1 patient had recurrent viral infections, and another patient had recurrent viral and bacterial infections. A pair of monozygotic twins with discordant phenotypes were included in the patient cohort [4]. In the 24 patients for whom photographs were available, dysmorphic features included a triangular-shaped face in 10; a high anterior hairline in 19; deep-set eyes in 5, almond-shaped eyes in 8; periorbital fullness in 6, thick nasal alae and flared nostrils in 9, full cheeks in 7, and a thin upper lip in 12. The degree of dysmorphism was variable.
INHERITANCE
The heterozygous mutations in 29 of the 34 patients with CAGS reported by Chopra et al. [1] occurred de novo. One patient inherited the disorder from affected parent, and another patient inherited it from a parent with low-level mosaicism for an ANKRD17 mutation. Parental inheritance was not determined in 3 patients.
Cytogenetic
In a patient (individual 6) with Chopra-Amiel-Gordon syndrome, Chopra et al. [1], identified a de novo heterozygous 1.16-Mb deletion encompassing 7 genes, including ANKRD17, by array CGH. The other 6 genes in the region were associated with an autosomal recessive pattern of disease inheritance when mutated (ADAMTS3, 605011; ALB, 103600; AFP, 104150) or were not known to be associated with a disease (COX18, 610428; AFM, 104150; RASSF6, 612620). Chopra et al. [1], therefore considered ANKRD17 to be the most likely candidate gene underlying the patient’s phenotype. Functional studies were not performed [3,4].
Molecular Genetics
In 34 patients from 32 families with Chopra-Amiel-Gordon syndrome, Chopra et al. [1] identified heterozygous mutations in the ANKRD17 gene (see, e.g., 615929.0001- 615929.0005). The mutations included 21 truncating or canonical splice site mutations, 9 missense mutations, 1 in-frame indel, and 1 microdeletion including ANKRD17 and 6 other genes. Molecular modeling suggested that most of the missense mutations disrupted the stability of ankyrin repeats as shown in Figure 3.
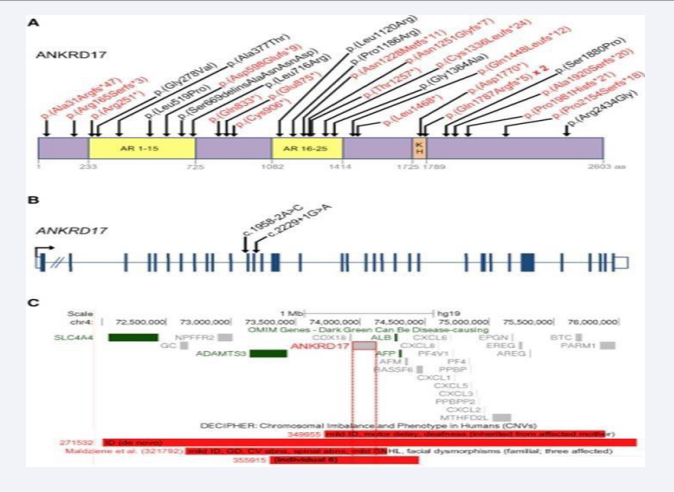
Figure 3: Distribution of pathogenic ANKRD17 variants and 4q13.3 deletions (A) Variants affecting coding sequence.
ACKNOWLEDGEMENTS
We extend our sincere thanks and gratitude to the medical staff specialized in the Scientific Committee for Muscular Dystrophy Diseases in Libya in general and the sub - committee in the south in particular.
Conflict of Interest
Coded as: OMIM:615929.0001,OMIM:615929.0002,OMIM:6 15929.0003,OMIM:615929.0004,OMIM:615929.0005,OMIM:61 9504,UMLS:C5561975.








































































































































