One Stone, Two Birds: Cyclindependent kinase 6 as a Therapeutic Target in T-cell Acute Lymphoblastic Leukemia
- 1. Department of Medicine, Molecular Oncology Research Institute, USA
- 2. Yale School of Medicine, USA
Abstract
Cyclin-dependent kinase 6 (CDK6), a D-type cyclin regulator, has aberrant expression in various kinds of malignancies, including T-cell acute lymphoblastic leukemia (T-ALL). This review highlights the importance of CDK6 as a common downstream mediator in both Notch- and PTENAKT-dependent T-cell malignancies. As a central molecular player linking extracellular signals to cell cycle machinery, CDK6 orchestrates network dynamics in normal versus cancerous cells. Albeit much remains to be exploited, CDK6 serves as a potential adjuvant therapeutic target in the Notch and PTEN-AKT subgroup of T-ALL.
Keywords
Cyclin D-dependent Kinase 6 (CDK6) , CDK6 kinase activity , T-cell lymphoblastic leukemia (T-ALL) , il2Ra (CD25) , Notch1 ,PTEN-AKT
Citation
Zou T, Hu JK, Daniele SG, Hu MG (2017) One Stone, Two Birds: Cyclin-dependent kinase 6 as a Therapeutic Target in T-cell Acute Lymphoblastic Leukemia. JSM Anat Physiol 2(2): 1015.
THE GENETICS AND MECHANISMS OF T-ALL
Acute lymphoblastic leukemia (ALL) is a quickly progressing cancer of white blood cells. The annual incidence of ALL in the US is roughly 4,000-3,000 of which afflict children. T-ALL accounts for about 15% and 25% in pediatric and adult ALLs, respectively. Pathophysiologically, T-ALL results from sustained activation of oncogenes and/or inactivation of tumor suppressors. Recent evidence indicates that more than 50% of all T-ALLs express a constitutively active Notch1 receptor [1], about 3% of cases harbor translocation mutation in CCND2 locus, resulting in aberrantly high levels of cyclin D2 expression, an activating partner of cyclin-dependent kinases (CDKs), which drives uncontrolled cell proliferation [2]. By contrast, 70% of T-ALL patients have a deletion mutation of the CDKN2A locus encoding the tumor suppressors p16INK4A and p14ARF [3,4], while approximately 15% of T-ALLs show chromosome deletions in 13q14.2, containing the retinoblastoma 1 (RB1) locus, encoding a retinoblastoma tumor suppressor protein (pRB) [5,6]. About 12% of ALLs display deletion mutation in CDKN1B, which encodes p27KIP1, another family inhibitor of CDKs [7]. Furthermore, 5% of T-ALLs have somatic mutations in the RUNX1 gene, which encodes a tumor suppressor and master regulator transcription factor with prominent roles in hematopoietic development [8-10]. And approximately 10-15% of T-ALLs show loss of PTEN tumor suppressor gene, a major negative regulator of AKT signaling [11,12]. Moreover, some T-ALL patients have heterozygous point mutations in the zinc finger DNA-binding protein domain of GATA3 [13], an important transcriptional regulator of T cell differentiation with an essential role in the development of early T cell progenitors [14], and a crucial tumor suppressor role in basal-like breast cancer [15].
CURRENT TREATMENT OF T-CELL ACUTE LYMPHOBLASTIC LEUKEMIA
In both children and adults, T-ALL is currently treated with a complex combination of chemotherapeutics to achieve sustained remission with low toxicity to normal cells in order to allow rapid hematological recovery for further treatment [16]. In addition, depletion of extracellular asparagine through the use of the enzyme L-asparaginase [17], in pediatric T-ALLs can prolong treatment effect. Hematopoietic cell transplantation has been used to treat adult T-ALL with high effectiveness [18]. Despite all the improved treatment protocols, a significant proportion of T-ALL patients suffer relapse, possibly due to the failure of current treatments in eradicating the most primitive leukemiainitiating cells or leukemia stem cells (LSCs), which initiate and sustain the disease in vivo. Treatment options for patients with relapsed or refractory T-ALL are limited, and include agents such as nelarabine and clofarabine, exhibiting efficacy in <20% of patients, or allogeneic stem cell transplantation. Moreover, all patients are clinically treated the same regardless of presenting symptoms since there are no available biomarkers for patient stratification. Thus, long-term toxicities of intensive therapy are becoming increasingly common. Taken together, all evidence underscores the need to develop more specific and highly effective anti-leukemic therapies.
THE ROLE OF CDK6 IN CELL CYCLE CONTROL
Cyclin-dependent kinases 4/6 (CDKs 4/6) play a wellestablished role in the regulation of the cell cycle in early G1 phase (Figure 1).
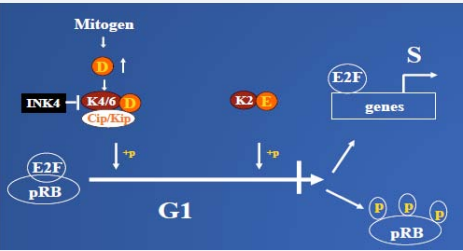
Figure 1: The role of CDK6 in normal cell cycle control: In the early G1 phase, D-type Cyclins are induced and assembled with, CDK4/6 (K4/ K6) in response to the mitogen stimulation. The kinases are activated and imported into nucleus. There, they start to phosphorylate one of their substrate pRB, inactivating its function. The complete inactivation of pRB also requires sequential phosphorylation by CDK2 (K2) kinase. Hyperphosphorylation of pRB results in releasing of transcription factors E2F, which transactivate a series of genes, required for G1-S transition. D-type CDKs play an important role in this process by linking the extracellular stimulation to the cell cycle machine. Overall, kinase activity is tightly regulated. Firstly, by definition, CDKs have to bind to cyclin to become active. Secondly, kinase activity is negatively regulated by binding to different CDK inhibitors such as the Ink4 family protein p16, and the Cip/Kip family proteins, p21 and p27. The kinase is also regulated by a series of phosphorylation and dephosphorylation steps. However, deregulation of any component in the signaling pathway can cause tumor formation
Proliferative stimuli induce the expression of D-type cyclins, which assemble with CDK4 and CDK6. This results in the activation of CDKs 4/6 and the nuclear import of their holoenzymes, where they collaborate with cyclin E-CDK2 to promote cell division by phosphorylation of pRB, as well as the pRB-related family proteins p107 and p130 [19]. Hyperphosphorylation of pRB results in activation or de-repression of E2F-dependent promoters and the transcription of genes required for S-phase entry [19,20]. Therefore, D-cyclin–CDK4/6 complexes play a catalytic role in the G1-S transition by negatively regulating pRB function. Apart from their well-documented CDKdependent role, D-cyclin–CDK4/6 complexes likely play a second, non-catalytic role in the G1-S transition by sequestering the Cip/ Kip family proteins p27KIP1 (p27) and p21CIP1 (p21) [21-23]. This mechanism helps to relieve cyclin E–CDK2 from its constraint, thereby facilitating its activation later in G1 phase. Hence, both pRB and p27 activity are negatively regulated by cyclin D-CDK4/6.
The Cip/Kip inhibitor family includes p21, p27 and p57KIP2 (p57) which associate with several different cyclin/CDK complexes through a conserved N-terminal domain that contains both cyclin and CDK binding sites [24-26]. Cip/Kip molecules inhibit CDK2 kinase activity and block cell cycle progression when they associate with CDK2. However, the function of CDK4/6- associated CIP/KIP proteins may be much more complex: there is strong evidence that Cip/Kip proteins serve as important assembly factors for D cyclin-CDK4/6 complexes, favoring cell cycle progression by a non-catalytic mechanism [27,28]. However, this function may not be universal because p27 and p21 stabilize but are not absolutely required for the formation of active D cyclin-CDK4 complexes, and both can inhibit the activity of D cyclin-CDK4 complexes under some circumstances [29].
Of the Cip/Kip family of CDK inhibitors, p21 and p57 are not significantly expressed in lymphoid organs, and mutations in their genes exhibit no effect on T-cell proliferation [30-32]. In contrast, targeted disruption of the murine p27 gene caused a pronounced lymphoid hyperplasia in the thymus and spleen, which was associated with increased T-lymphocyte proliferation. However, the development of these cells was remarkably unperturbed [33-35]. Furthermore, p27 also governs CDK activity during the transition from quiescent G1-phase to S-phase in T-lymphocytes [36-38]. Recent evidence shows that about 12% of ALLs display a deletion mutation in the p27 gene, CDKN1B (57). Therefore, p27 plays a key role in the negative modulation of T-cell proliferation.
A second set of CDK inhibitors (CKIs) also acts to restrain proliferation in most cell types. In contrast to the Cip/Kip family, the INK family of CKIs including p15INK4b (p15), p16 INK4a (p16), p18 INK4c (p18), and p19 INK4d (p19) can independently associate with CDK4/6. The stable CDK-INK4 complexes inactivate CDKs by preventing their association with the activating cyclin D subunit [39-41]. Among the four INK4 CDK inhibitors, inactivation of p15 or p19 was recently shown to have little or no significant effect on T-cell proliferation [42,43]. In contrast, 70% of T-ALL patients have a deletion mutation of the tumor suppressors p16INK4A and p14ARF [3,4]; loss of p18 alone in T-cells leads to hyperproliferation and development of lymphoproliferative disorder and T-cell lymphomas [44,45], despite high expression of both p18 and p19 in lymphoid organs/cells [30,46,47]. Furthermore, biochemical analysis of the G1 regulatory proteins indicates that p18 is stable and preferentially inhibitory to CDK6 but not CDK4 activity in activated T cells [45]. Thus, the INK family of CKIs is an important class of inhibitory molecules that modulate T-cell development and tumorigenesis.
Since their discovery, CDK4/6 have been widely thought to be important for initiation of the cell cycle in response to growth regulatory signals and to regulate the cell cycle progressionthrough the phosphorylation of pRB [20,48-51]. In some tissues, preferential expression of CDK4 or CDK6 supports the idea that these kinase subunits may have redundant functions. For instance, CDK4 is highly expressed in mesenchymal cell types such as fibroblasts and osteoblasts. In contrast, CDK6 is predominant in all hematopoietic cell types [51-53]. Consistent with this notion, CDK6 is the earliest CDK to be induced during human T-cell activation [54,55], and high-level expression of pre-activated CDK6 were found in CD8+ memory cells, mediating rapid division after antigen re-stimulation [56]. Importantly, recent analyses of CDK6 knockout animals reported significantly decreased thymic cellularity, with unappreciable alteration in other tissues, indicating the specificity of CDK6 in the thymus [57-59]. Nevertheless, most tissues, including those listed above, express both CDK4 and CDK6 at detectable levels, perhaps indicating that these subunits have discrete, non-overlapping functions in certain cell types or during specific developmental stages.
ONCOGENIC CDK6 IN T-ALL
Consistent with the idea that CDK4 and CDK6 may differentially impact proliferation in a cell-type dependent manner, CDK6 is over expressed in human T-cell Lymphoblastic Lymphoma/ALL [52,60-62], and the CDK6 locus is amplified in 25% of peripheral T-cell lymphomas [61]. CDK6 also plays a role in the development of lymphoid malignancy in E47-deficient T-cell lineages [62]. Alternatively, aberration in cell cycle regulation, especially sustained CDK4/6 activation, is a common theme in the majority of human cancers including T-ALLs [63,64]; for instance, 70% of T-ALL patients have deletion mutation of the tumor suppressors p16INK4A and p14ARF, as mentioned above. In contrast, CDK4 is specifically mutated in human melanomas [65,66], and overexpressed in a significant fraction of human breast cancers [67,68]. Moreover, in the past few years, several groups have demonstrated that CDK6 has a specific role in differentiation of certain cell types such as in astrocytes [69], osteoblasts [70], and murine erythroid leukemia cells [71]. This function is apparently not shared with CDK4.
ONCOGENIC SIGNALING PATHWAYS IN CDK6- MEDIATED T-ALL
Upstream regulators of CDK6
The oncogenic role of CDK6 in T-cell is well documented in genetic models. For example, we and other groups have provided genetic and molecular evidence to demonstrate that CDK6 kinase activity is critical to induction of Notch1-mediated T-ALL in a mouse model [64,72,73]. We found that donor cells lacking the CDK6 protein or its kinase activity, while expressing intact CDK4, are resistant to transformation by activated Notch1 as a consequence of reduced proliferation and increased apoptosis. Re-expression of CDK6 in CDK6-deficient or kinase-dead stem cells rescues this defect, arguing for a cell-autonomous effect of CDK6 downstream of Notch1 in leukemogenesis. In complementary studies, Choi, YJ et al. [73], and Sawai CM et al. [64], have shown that deletion of cyclin D3 (an activating partner of CDK4/6) or treatment with palbociclib (PD0332991), an inhibitor of CDK4/6, induced cell cycle arrest and apoptosis in Notch1-induced mouse T-ALLs and prolonged survival of mice bearing these leukemia [64,73].Moreover, we have also extended our findings to human leukemia by testing the response of human T-ALL cell lines with constitutively active Notch1 mutations (1) to PD0332991 or Cdk6-shRNAs. PD0332991 or Cdk6-shRNAs treatment inhibited cell proliferation and induced apoptosis, recapitulating the effects seen in the mouse models. Taken together, these data indicate that CDK6, as a downstream effector of Notch1 [58,59,72], is required for initiation and maintenance of Notch1-induced T-ALL.
Recent evidence also indicates that AKT-activating mutations are frequently found in many types of human T-ALL and murine T–cell tumors [74-77]. Over-expression of an active form of AKT (MyrAkt1) in WT T-cell progenitors induces T–cell lymphoma accompanied with increased level of CD44 [58,78]. However, CDK6 deficient mice are resistant to MyrAkt1-induced T-cell hematopoietic malignancies, in support of CDK6 being a major oncogenic driver downstream of AKT, with higher expression of CD25 and lower expression of CD44 [58]. Thus, CDK6 is also required for T-cell tumorigenesis in an AKT-dependent pathway.
In conclusion, CDK6 acts as a common downstream target of Notch and PI3K-AKT signaling pathways in T-cell tumorigenesis, which imply that CDK6 can be a promising novel and highly selective treatment for T-ALLs.
DOWNSTREAM EFFECTORS OF CDK6: CD25
CD25, the alpha chain of interleukin-2 (IL-2) receptor (IL2Ra), is expressed on the surface of certain immune cells, such as lymphocytes. The role of IL2 and its receptor is well known for their functions in the immune system, specifically in mediating tolerance and immunity, which occurs through their direct effect on T-cells. In the human and murine thymus, IL-2/IL-2R promotes the differentiation of regulatory T-cells to prevent autoimmune disease [79], they also promote the differentiation of effector and memory T-cells to fight off infections [79], and to maintain cell-mediated immunity [79,80]. However, the role of CD25 in tumorigenesis remains elusive.
In a different disease context, PIM kinase inhibitors target CD25-positive acute myeloid leukemia (AML) cells through the suppression of STAT5 activation, leading to inhibition of AML cell proliferation. Thus CD25, a STAT5 regulated gene, can potentially be used as a biological marker for effective response to PIMinhibitor treatment in AML [81]. Moreover, CD25 is the marker for diagnosis of adult T-cell lymphoma/leukemia and hairy cell leukemia [82]. By contrast, Willerford, DM et al., observed that CD25 deficient mice developed polyclonal T- and B-cell expansion leading to enlargement of lymphoid tissue [83], suggesting that CD25 has anti-proliferative effect on lymphocytes. More studies revealed that CD25 balances clonal proliferation and cell death to ensure the appropriate size and function of lymphocytes by regulating Fas-dependent T-cell death [84]. Consistent with this observation, loss of CDK6 or its kinase activity causes reduced cell numbers of stem cells and thymocytes, along with increased CD25 expression [58,59]. Similarly, CD25 is increased in the mouse leukemia cells treated with CDK4/6 inhibitor, indicating that CDK6 activity suppresses CD25 expression in the presence of constitutively active Notch1 [72]. Furthermore, re-expression of CDK6 in knockout (KO) or kinase-dead (K43M) stem/progenitor(LKs) cells dramatically reduced CD25 level in leukemic cells and restored T-ALL [72]. By contrast, ablation of CD25 in K43M mice (Cd25–/–;K43M) reverses the protective effects K43M LKs exhibits in the development and transformation of leukemia cells, demonstrating the essential role of CD25 in CDK6-mediated, Notch-induced transformation [72]. Taken together, the above findings argue that CD25 could display a novel tumor suppressor function in this genetic context. Our data revealed a previously unknown relationship between CD25 repression and CDK6 expression in T-cell development and T-ALL tumorigenesis.
MEDIATORS LINKING CDK6 AND CD25
Foxp3 directly regulates CD25 expression [85,86]. However, although attenuated, CD25 is still highly expressed in Foxp3- deficient Treg cells [87], suggesting that additional transcription factors or pathways are involved in CD25 regulation. The bestcharacterized targets of CDK6 are pRB [19,20], and RUNX1, a tumor suppressor in T-ALLs [88,89]. Phosphorylation of pRB or RUNX1 by CDK6 results in loss of function of pRB [19,20], or increased degradation of RUNX1 [90,91]. It is possible that pRB and/or Runx1 are mediating CDK6 function in regulating CD25 expression, and thus T-cell development and tumorigenesis. However, ablation of pRB or RUNX1 on K43M background does not alter CD25 expression nor does it rescue the defects in Notchinduced leukemogenesis associated with the loss of CDK6 kinase activity; although deletion of pRB, but not RUNX1, partially restores the development of K43M thymocytes [72], suggesting that pRB and RUNX1 are not downstream mediators of CDK6 in Notch1-induced mouse T-ALL.
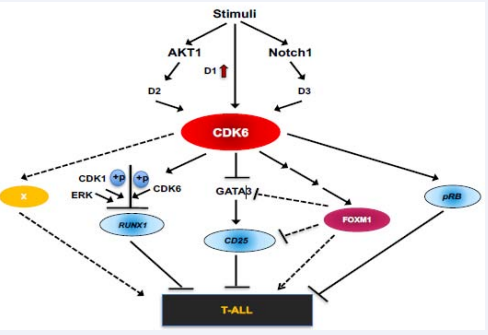
Figure 2: A working model of the role of CDK6 in T-ALL: In response to stimuli, increased expression of cyclin D1 and CDK6 leads to increased CDK6 activation, while Notch1 and AKT1 are also independently activated in parallel. Notch1 further activates CDK6 via upregulation of CDK6 and/ or by increased cyclin D3 protein, while AKT1 activates CDK6 through the stabilization of cyclin D2. Once activated, CDK6 can phosphorylate pRB, resulting in its inactivation. CDK6, along with ERK and CDK1 can also phosphorylate RUNX1 thereby promoting RUNX1 proteolytic degradation. On a different molecular path, phosphorylation of FOXM1 by CDK6 stabilizes FOXM1, which in turn promotes methylation of the GATA3 promoter, decreasing GATA3 expression and the subsequent recruitment to the CD25 proximal promoter region. CD25 expression is consequently reduced and T-ALL develops. Devoid of CDK6 protein/kinase activity, pRB and RUNX1 remain active, which suppress the tumorigenesis in CD25-independent manner. Contrastingly, without CDK6, FOXM1 is in its inactive state, leading to CD25 upregulation. Overall, T-ALL is suppressed by FOXM1 inactivity, and by increasing RUNX1, pRB, GATA3, and CD25 expression or activity
Another potential effector of CD25 expression downstream of CDK6 is FOXM1, widely known as a transcriptional activator involving in G1-S transition and senescence suppression [92]. Phosphorylation of FOXM1 diverts its proteasomal degradation, leading to the stabilization of FOXM1 protein [92]. Consistent with this finding, we confirmed that deficiency or inhibition of CDK6 kinase activity is associated with decreased FOXM1 protein levels in thymocytes or in human T-ALL cell lines [72]. It is conceivable that FOXM1 may negatively regulate T-ALL by promoting methylation of the GATA3 promoter, as is the case in breast cancer [93], leading to a reduction of GATA3 and CD25, thereby facilitating the development of T-ALL.
As such, GATA 3 could be another potential effector of CD25 expression downstream of CDK6. We showed previously that expression of GATA3 is higher in KO/K43M thymocytes but lower in WT/R31C cells [59], and GATA3 knockdown in differentiated T cells correlated with a robust reduction of CD25 levels [59]. Moreover, there are GATA factor consensus binding sequences within 1.3 kb and 4.0 kb of the transcriptional start sites of mouse [94,95], and human [95-97], IL2Ra genes, respectively. Therefore, it is possible that CDK6 regulates CD25 expression in part through repression of GATA3.
Based on our published findings, we propose that CDK6 is integrated into the T-ALL pathways as follows (Figure 2). In response to stimuli, increased expression of cyclin D1 [98], and CDK6 [52,60-62], leads to increased activation of CDK6. The canonical cascade of events is initiated, including the activation of Notch1 and AKT1 [100]. Notch1 activates CDK6 via upregulation of CDK6 and/or by increased cyclin D3 protein, while AKT1 activates CDK6 by stabilizing cyclin D2 [102]. Once activated, CDK6 initiates stem cell transformation via a CD25-dependentor -independent manner. Both pRB and RUNX1 suppress T-ALL via CD25-independent pathway, for instance, loss of RUNX1 may contribute to the pathogenesis of T-ALL, perhaps via decreased expression of PKCq and reactive oxygen species [103,104]. However, phosphorylation of pRB by CDK6 results in inactivation of pRB [19,20], and phosphorylation of RUNX1 by CDK6 and other kinases such as ERK and CDK1 [88], promotes RUNX1 proteolytic degradation [88], resulting in a reduction of RUNX1. On the other hand, phosphorylation of FOXM1 by CDK6 stabilizes FOXM1, which in turn promotes methylation of the GATA3 promoter and decreases GATA3 expression and its subsequent recruitment to the proximal promoter regions of the CD25 locus, IL2Ra. Consequently, CD25 expression is reduced, leading to T-ALL development. In the absence of CDK6 protein/ kinase activity, pRB and RUNX1 are active, which suppress tumorigenesis in a CD25-independent manner. Likewise, if CDK6 activity were abolished, FOXM1 remains inactive, while CD25 is up-regulated. Together, through the inhibition of FOXM1 activity in G1-S transition and senescence suppression, and by increasing the levels or activity of RUNX1, pRB, GATA3, and CD25, T-ALL is suppressed.
In summary, CDK6 kinase activity is required for T-ALL initiation and maintenance. As such, CDK4/6 inhibition would not only block cell proliferation, induces apoptosis, but also upregulate CD25 expression. Our studies suggest that while CDK6 can be a therapeutic target in human T-ALL, the expression of CD25 could serve as a valuable biomarker for predicting the clinical response to CDK6 inhibitor treatment in T-ALL patients.
ACKNOWLEDGEMENTS
This work was supported by a V Foundation Translational Research Grant, Tufts Medical Center Research Fund, Tufts CTSI-Catalyst Award (UL1 TR001064), Moore/Moreau Cancer Research Project Grant, and Tufts University Seed Grants to M.G.H.
REFERENCES
- Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004; 306: 269-271.
- Clappier E, Cuccuini W, Cayuela JM, Vecchione D, Baruchel A, Dombret H, et al. Cyclin D2 dysregulation by chromosomal translocations to TCR loci in T-cell acute lymphoblastic leukemias. Leukemia. 2006; 20: 82-86.
- Hebert J, Cayuela JM, Berkeley J, Sigaux F. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood. 1994; 84: 4038-4044.
- Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002; 1:75-87
- Van Vlierberghe P, Ambesi-Impiombato A, De Keersmaecker K, Hadler M, Paietta E, Tallman MS, et al. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood. 2013; 122: 74-82.
- Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukemia. Nature. 2007; 446:758-764.
- Remke M, Pfister S, Kox C, Toedt G, Becker N, Benner A, et al. High-resolution genomic profiling of childhood T-ALL reveals frequent copy-number alterations affecting the TGF-beta and PI3K-AKT pathways and deletions at 6q15-16.1 as a genomic marker for unfavorable early treatment response. Blood. 2009; 114: 1053-1062.
- Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996; 84: 321-330.
- Cai Z, de Bruijn M, Ma X, Dortland B, Luteijn T, Downing RJ, et al. Haploinsufficiency of AML1 affects the temporal and spatial generation of hematopoietic stem cells in the mouse embryo. Immunity. 2000; 13: 423-431.
- Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005; 106: 494-504.
- Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007; 13: 1203-1210.
- Mendes RD, Sarmento LM, Cante-Barrett K, Zuurbier L, Buijs-Gladdines JG, Povoa V, et al. PTEN microdeletions in T-cell acute lymphoblastic leukemia are caused by illegitimate RAG-mediated recombination events. Blood. 2014; 124: 567-578.
- Zahirieh A, Nesbit MA, Ali A, Wang K, He N, Stangou M, et al. Functional analysis of a novel GATA3 mutation in a family with the hypoparathyroidism, deafness, and renal dysplasia syndrome. J Clin Endocrinol Metab. 2005; 90: 2445-2450.
- Ting CN, Olson MC, Barton KP, Leiden JM. Transcription factor GATA-3 is required for development of the T-cell lineage. Nature. 1996; 384: 474-478.
- Chou J, Provot S, Werb Z. GATA3 in development and cancer differentiation: cells GATA have it! J Cell Physiol. 2010; 222: 42-49.
- Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood. 2017; 129: 1134-1142.
- Amylon MD, Shuster J, Pullen J, Berard C, Link MP, Wharam M, et al. Intensive high-dose asparaginase consolidation improves survival for pediatric patients with T cell acute lymphoblastic leukemia and advanced stage lymphoblastic lymphoma: a Pediatric Oncology Group study. Leukemia. 1999; 13: 335-342.
- Goldstone AH, Richards SM, Lazarus HM, Tallman MS, Buck G, Fielding AK, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood. 2008; 111: 1827-1833.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999; 13: 1501-1512.
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995; 81: 323-330.
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994; 8: 9-22.
- Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995; 9: 1831-1845.
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995; 9: 1149-1163.
- Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994; 78: 67-74.
- Chen J, Jackson PK, Kirschner MW, Dutta A. Separate domains of p21 involved in the inhibition of Cdk kinase and PCNA. Nature. 1995; 374: 386-388.
- Luo Y, Hurwitz J, Massague J. Cell-cycle inhibition by independent CDK and PCNA binding domains in p21Cip1. Nature. 1995; 375: 159-161.
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997; 11: 847-862.
- Zhang H, Hannon GJ, Beach D. p21-containing cyclin kinases exist in both active and inactive states. Genes Dev. 1994; 8: 1750-1758.
- Bagui TK, Mohapatra S, Haura E, Pledger WJ. P27Kip1 and p21Cip1 are not required for the formation of active D cyclin-cdk4 complexes. Mol Cell Biol. 2003; 23: 7285-7290.
- Guan KL, Jenkins CW, Li Y, Nichols MA, Wu X, O'Keefe CL, et al. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 1994; 8: 2939-2952.
- Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, et al. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995; 9: 650-662.
- Parker SB, Eichele G, Zhang P, Rawls A, Sands AT, Bradley A, et al. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science. 1995; 267: 1024-1027.
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996; 85: 733-744.
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell. 1996; 85: 721-732.
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996; 85: 707-720.
- Boussiotis VA, Freeman GJ, Taylor PA, Berezovskaya A, Grass I, Blazar BR, et al. p27kip1 functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nat Med. 2000; 6: 290-297.
- Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH, et al. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994; 372: 570-573.
- Powell JD, Bruniquel D, Schwartz RH. TCR engagement in the absence of cell cycle progression leads to T cell anergy independent of p27 (Kip1). Eur J Immunol. 2001; 31: 3737-3746.
- Brotherton DH, Dhanaraj V, Wick S, Brizuela L, Domaille PJ, Volyanik E, et al. Crystal structure of the complex of the cyclin D-dependent kinase Cdk6 bound to the cell-cycle inhibitor p19INK4d. Nature. 1998; 395: 244-250.
- Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature. 1998; 395: 237-243.
- Peter M, Herskowitz I. Joining the complex: cyclin-dependent kinase inhibitory proteins and the cell cycle. Cell. 1994; 79: 181-184.
- Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin-Caballero J, et al. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. Embo J. 2000; 19: 3496-3506.
- Zindy F, van Deursen J, Grosveld G, Sherr CJ, Roussel MF. INK4d-deficient mice are fertile despite testicular atrophy. Mol Cell Biol. 2000; 20: 372-378.
- Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998; 12: 2899-2911.
- Kovalev GI, Franklin DS, Coffield VM, Xiong Y, Su L. An important role of CDK inhibitor p18 (INK4c) in modulating antigen receptor-mediated T cell proliferation. J Immunol. 2001; 167: 3285-3292.
- Chan FK, Zhang J, Cheng L, Shapiro DN, Winoto A. Identification of human and mouse p19, a novel CDK4 and CDK6 inhibitor with homology to p16ink4. Mol Cell Biol. 1995; 15: 2682-2688.
- Hirai H, Roussel MF, Kato JY, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995; 15: 2672-2681.
- Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992; 71: 323-334.
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994; 14: 2066-2076.
- Meyerson M, Enders GH, Wu CL, Su LK, Gorka C, Nelson C, et al. A family of human cdc2-related protein kinases. Embo J. 1992; 11: 2909-2917.
- Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994; 14: 2077-2086.
- Chilosi M, Doglioni C, Yan Z, Lestani M, Menestrina F, Sorio C, et al. Differential expression of cyclin-dependent kinase 6 in cortical thymocytes and T-cell lymphoblastic lymphoma/leukemia. Am J Pathol. 1998; 152: 209-217.
- Della Ragione F, Borriello A, Mastropietro S, Della Pietra V, Monno F, Gabutti V, et al. Expression of G1-phase cell cycle genes during hematopoietic lineage. Biochem Biophys Res Commun. 1997; 231: 73-76.
- Modiano JF, Domenico J, Szepesi A, Terada N, Lucas JJ, Gelfand EW. Symmetry of the activation of cyclin-dependent kinases in mitogen and growth factor-stimulated T lymphocytes. Ann N Y Acad Sci. 1995; 766: 134-148.
- Nagasawa M, Melamed I, Kupfer A, Gelfand EW, Lucas JJ. Rapid nuclear translocation and increased activity of cyclin-dependent kinase 6 after T cell activation. J Immunol. 1997; 158: 5146-5154.
- Veiga-Fernandes H, Rocha B. High expression of active CDK6 in the cytoplasm of CD8 memory cells favors rapid division. Nat Immunol. 2004; 5: 31-37.
- Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004; 118: 493-504.
- Hu MG, Deshpande A, Enos M, Mao D, Hinds EA, Hu GF, et al. A requirement for cyclin-dependent kinase 6 in thymocyte development and tumorigenesis. Cancer Res. 2009; 69: 810-818.
- Hu MG, Deshpande A, Schlichting N, Hinds EA, Mao C, Dose M, et al. CDK6 kinase activity is required for thymocyte development. Blood. 2011; 117: 6120-6131.
- Lien HC, Lin CW, Huang PH, Chang ML, Hsu SM. Expression of cyclin-dependent kinase 6 (cdk6) and frequent loss of CD44 in nasal-nasopharyngeal NK/T-cell lymphomas: comparison with CD56-negative peripheral T-cell lymphomas. Lab Invest. 2000; 80: 893-900.
- Nagel S, Leich E, Quentmeier H, Meyer C, Kaufmann M, Drexler HG, et al. Amplification at 7q22 targets cyclin-dependent kinase 6 in T-cell lymphoma. Leukemia. 2008; 22: 387-392.
- Schwartz R, Engel I, Fallahi-Sichani M, Petrie HT, Murre C. Gene expression patterns define novel roles for E47 in cell cycle progression, cytokine-mediated signaling, and T lineage development. Proc Natl Acad Sci U S A. 2006; 103: 9976-9981.
- O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016; 13: 417-430.
- Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz JC, Strikoudis A, et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell. 2012; 22: 452-465.
- Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996; 68: 67-108.
- Zuo L, Weger J, Yang Q, Goldstein AM, Tucker MA, Walker GJ, et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996; 12: 97-99.
- An HX, Beckmann MW, Reifenberger G, Bender HG, Niederacher D. Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation. Am J Pathol. 1999; 154: 113-118.
- Samady L, Dennis J, Budhram-Mahadeo V, Latchman DS. Activation of CDK4 gene expression in human breast cancer cells by the Brn-3b POU family transcription factor. Cancer Biol Ther. 2004; 3: 317-323.
- Ericson KK, Krull D, Slomiany P, Grossel MJ. Expression of cyclin-dependent kinase 6, but not cyclin-dependent kinase 4, alters morphology of cultured mouse astrocytes. Mol Cancer Res. 2003; 1: 654-664.
- Ogasawara T, Kawaguchi H, Jinno S, Hoshi K, Itaka K, Takato T, et al. Bone morphogenetic protein 2-induced osteoblast differentiation requires Smad-mediated down-regulation of Cdk6. Mol Cell Biol. 2004; 24: 6560-6568.
- Matushansky I, Radparvar F, Skoultchi AI. Reprogramming leukemic cells to terminal differentiation by inhibiting specific cyclin-dependent kinases in G1. Proc Natl Acad Sci U S A. 2000; 97: 14317-14322.
- Jena N, Sheng J, Hu JK, Li W, Zhou W, Lee G, et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-cell acute lymphoblastic leukemia. Leukemia. 2016; 30: 1033-1043.
- Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012; 22: 438-451.
- Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002; 14: 381-395.
- Zweidler-McKay PA, Pear WS. Notch and T cell malignancy. Semin Cancer Biol. 2004; 14: 329-340.
- Malstrom S, Tili E, Kappes D, Ceci JD, Tsichlis PN. Tumor induction by an Lck-MyrAkt transgene is delayed by mechanisms controlling the size of the thymus. Proc Natl Acad Sci U S A. 2001; 98: 14967-14972.
- Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009; 114: 647-650.
- Mao C, Tili EG, Dose M, Haks MC, Bear SE, Maroulakou I, et al. Unequal contribution of Akt isoforms in the double-negative to double-positive thymocyte transition. J Immunol. 2007; 178: 5443-5453.
- Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol. 2011; 23: 598-604.
- Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity. 2010; 33: 153-165.
- Guo Z, Wang A, Zhang W, Levit M, Gao Q, Barberis C, et al. PIM inhibitors target CD25-positive AML cells through concomitant suppression of STAT5 activation and degradation of MYC oncogene. Blood. 2014; 124: 1777-1789.
- Mais D. Quick Compendium of Clinical Pathology. 3rd edition. Am Soc Clin Pathol. 2014.
- Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995; 3: 521-530.
- Lynch DH, Ramsdell F, Alderson MR. Fas and Fas L in the homeostatic regulation of immune responses. Immunol Today. 1995; 16: 569-574.
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003; 299: 1057-1061.
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003; 4: 330-336.
- Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, et al. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007; 8: 359-368.
- Biggs JR, Peterson LF, Zhang Y, Kraft AS, Zhang DE. AML1/RUNX1 phosphorylation by cyclin-dependent kinases regulates the degradation of AML1/RUNX1 by the anaphase-promoting complex. Mol Cell Biol. 2006; 26: 7420-7429.
- Zhang L, Fried FB, Guo H, Friedman AD. Cyclin-dependent kinase phosphorylation of RUNX1/AML1 on 3 sites increases transactivation potency and stimulates cell proliferation. Blood. 2008; 111: 1193-1200.
- Della Gatta G, Palomero T, Perez-Garcia A, Ambesi-Impiombato A, Bansal M, Carpenter ZW, et al. Reverse engineering of TLX oncogenic transcriptional networks identifies RUNX1 as tumor suppressor in T-ALL. Nat Med. 2012; 18: 436-440.
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012; 481: 157-163.
- Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011; 20: 620-634.
- Carr JR, Kiefer MM, Park HJ, Li J, Wang Z, Fontanarosa J, et al. FoxM1 regulates mammary luminal cell fate. Cell Rep. 2012; 1: 715-729.
- Sperisen P, Wang SM, Soldaini E, Pla M, Rusterholz C, Bucher P, et al. Mouse interleukin-2 receptor alpha gene expression. Interleukin-1 and interleukin-2 control transcription via distinct cis-acting elements. J Biol Chem. 1995; 270: 10743-10753.
- Bucher P, Corthesy P, Imbert J, Nabholz M. A conserved IL-2 responsive enhancer in the IL-2R alpha gene. Immunobiol. 1997; 198: 136-143.
- John S, Robbins CM, Leonard WJ. An IL-2 response element in the human IL-2 receptor alpha chain promoter is a composite element that binds Stat5, Elf-1, HMG-I(Y) and a GATA family protein. Embo J. 1996; 15: 5627-5635.
- Lecine P, Algarte M, Rameil P, Beadling C, Bucher P, Nabholz M, et al. Elf-1 and Stat5 bind to a critical element in a new enhancer of the human interleukin-2 receptor alpha gene. Mol Cell Biol. 1996; 16: 6829-6840.
- Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014; 510: 547-551.
- Fukuda D, Aikawa E, Swirski FK, Novobrantseva TI, Kotelianski V, Gorgun CZ, et al. Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc Natl Acad Sci U S A. 2012; 109: E1868-1877.
- Ribeiro DL, Goes RM, Pinto-Fochi ME, Taboga SR, Abrahamsson PA, Dizeyi N. AKT and AMPK activation after high-fat and high-glucose in vitro treatment of prostate epithelial cells. Horm Metab Res. 2014; 46: 471-476.
- Joshi I, Minter LM, Telfer J, Demarest RM, Capobianco AJ, Aster JC, et al. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood. 2009; 113: 1689-1698.
- Muise-Helmericks RC, Grimes HL, Bellacosa A, Malstrom SE, Tsichlis PN, Rosen N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem. 1998; 273: 29864-29872.
- Giambra V, Jenkins CR, Wang H, Lam SH, Shevchuk OO, Nemirovsky O, et al. NOTCH1 promotes T cell leukemia-initiating activity by RUNX-mediated regulation of PKC-theta and reactive oxygen species. Nat Med. 2012; 18: 1693-1698.








































































































































