An open-label, balanced, randomized, two treatments, two sequences, four periods, fully replicate, crossover study to evaluate the bioequivalence of Dasatinib 100 mg film-coated tablets of Abbott Laboratories versus Sprycel
- 1. AZIDUS Laboratories Ltd.
ABSTRACT
PRYCEL (dasatinib) is a kinase inhibitor used in the treatment of Newly diagnosed Philadelphia chromosome positive (Ph+) chronic myelogenous leukaemia (CML) in the chronic phase, Chronic, accelerated, or myeloid or lymphoid blast phase Ph+ CML with resistance or intolerance to prior therapy including imatinib and Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ALL) with resistance or intolerance to prior therapy. The purpose of this study was to evaluate the bioequivalence between Dasatinib 100 mg filmcoated tablets of Abbott Laboratories versus Sprycel® (Dasatinib) 100 mg film-coated tablets of Bristol-Myers Squibb under both fasting and fed condition in healthy subjects. An open-label, balanced, randomized, two treatments, two sequences, four periods, fully replicate, crossover single dose study with washout period of 02 days under both fasting and fed condition was carried out in 40 subjects in the age group of 22 to 43 years and 32 subjects in fasting and 37 subjects in fed study completed all four periods of the study. The pharmacokinetic samples collected from subjects who completed the study were analysed to determine the plasma concentration of Dasatinib using a validated bio-analytical method.
In the fasting condition, the ISCV of Cmax for reference product was calculated to be 104.94%, hence, 90% confidence interval limit was widened using scaled-averagebioequivalence and the value obtained 72.44% - 123.72% was found to be within the widened limits of 69.84% to 143.19%.
90% confidence interval of AUC0-t was 84.84% - 116.94%, which was within the acceptable limits of 80.00% to 125.00%.
In the fed condition, 90% confidence interval of Cmax and AUC0-t were 102.34% - 120.53% and 100.72% - 112.80% respectively, which were found to be within the acceptable Limits.
KEYWORDS
• Dasatinib
• Sprycel
• film coated tablets
• Bioavailability
• Bioequivalence
• Pharmacokinetics
CITATION
Arjun AO, Srinivas G, Gavino-Gutierrez AM, Perez-Perez MP, Hurtado-Colorado K. ABBOTT (2022) An open-label, balanced, randomized, two treatments, two sequences, four periods, fully replicate, crossover study to evaluate the bioequivalence of Dasatinib 100 mg film-coated tablets of Abbott Laboratories versus Sprycel® (Dasatinib) 100 mg film-coated tablets of Bristol-Myers Squibb under both fasting and fed condition in healthy subjects. JSM Bioequiv Bioavailab 2(1): 1007.
ABBREVIATIONS
AUC: Area under the concentration versus time curve; AUC0-t: Area under the plasma concentration versus time curve from zero to time t; BMI: Body Mass Index; CDSCO: Central Drugs Standard Control Organization; Cmax: Concentration Maximum; CV: Coefficient of Variation; IEC: Independent Ethics Committee; ISCV: Intra-subject Co-efficient of Variation; LCMS/MS: Liquid Chromatography Tandem Mass Spectrometry; mg: Milligram; mL: Millilitre, mM: Milli Molar; ng/mL: Nanogram per millilitre; PK: Pharmacokinetic; Tmax: Time taken to reach maximum concentration.
INTRODUCTION
Bioavailability study: A bioavailability study is done by estimating the concentration of the drug in the plasma or blood after administration of drug.
Bioequivalence study: Bioequivalence study is used to assess the in vivo biological equivalence of a drug manufactured by two different manufacturers.
Many drug patents have recently expired or are scheduled to expire in the near future. In response, many drug manufacturers have expanded their generic drug profile, which requires them to conduct clinical trials that demonstrate that their generic equivalents perform similarly to the innovator drug product Regulations introduced by the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) over the last thirty-five years have strengthened measures to ensure the bioequivalence of drug products, which may be simultaneously manufactured by multiple drug makers. Bioequivalence and bioavailability testing standards have also emerged following recognition that bioequivalence and variations in the bioavailability of drug products can result in therapeutic failure and/or toxicity. SPRYCEL (dasatinib) is a kinase inhibitor. Dasatinib is N-(2-chloro-6-m ethylphenyl)-2- [[6-[4-(2-hydroxyethyl)-1-piperazinyl ]-2-methyl-4-pyrimidinyl] amino ]-5-thiazolecarboxamide, monohydrate chemically. Its molecular formula is C22H26C1 N7 O2 S • H2 0, and its molecular weight is 506.02. Dasatinib is a white to off-white powder. The drug substance is insoluble in water and slightly soluble in ethanol and methanol.
This study was designed to evaluate the relative bioavailability of the test product Dasatinib 100 mg film-coated tablets of Abbott Laboratories and reference product Sprycel® (Dasatinib) 100 mg film-coated tablets of Bristol-Myers Squibb under both fasting and fed condition in healthy subjects.
MATERIALS AND METHODS
Materials
Test product, dose and mode of administration: Dasatinib 100 mg film-coated tablets, 01 x 100 mg, Oral with 240 mL of water in sitting posture under both fasting and fed condition, Lot #: 20H268.
Reference product, dose and mode of administration: Sprycel® (Dasatinib) 100 mg film-coated tablets, 01 x 100 mg, Oral with 240 mL of water in sitting posture under both fasting and fed condition, Lot # MA0527.(Figure 1-4)
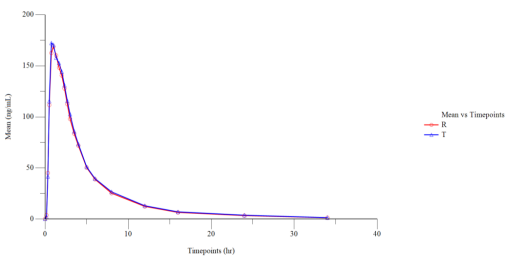
Figure 1 Linear Plot of Mean Plasmatic Dasatinib (Fasting) Concentration vs. Time Points (N=32).
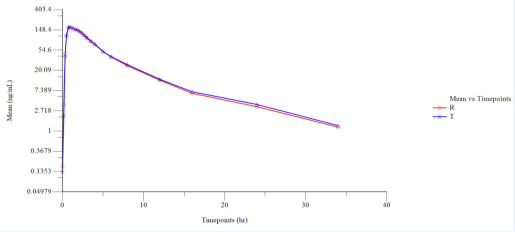
Figure 2 Semi log Plot of Mean Plasmatic Dasatinib (Fasting) Concentration vs. Time Points (N=32).
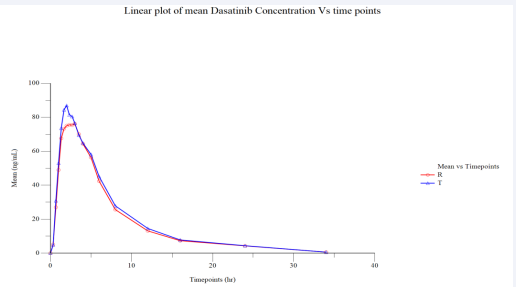
Figure 3 Linear Plot of Mean Plasmatic Dasatinib (Fed) Concentration vs. Time Points (N=37).
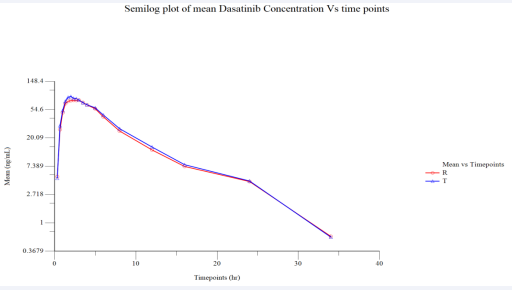
Figure 4 Semi log Plot of Mean Plasmatic Dasatinib (Fed) Concentration vs. Time Points (N=37).
Methodology
The study protocol with annexes was prepared and IEC approval was obtained before initiation of the study. Study subjects were screened and enrolled in the study as per the IEC approved protocol. Written informed consent was obtained from each volunteer for screening prior to initiation of screening procedure and for the study prior to enrolment. Individual counselling was then given to the willing volunteers by the Investigator in private and any questions and concerns were addressed prior to obtaining consent. The Principal investigator/ sub-investigator/physician reviewed all the screening results to assess eligibility of each volunteer. Subjects were enrolled in the study based on the inclusion and exclusion criteria. Forty subjects in the range of age between 22 to 43 years, who met the study eligibility criteria, participated in the study.
This study was designed based on the known pharmacokinetic profile of the investigational product and general accepted standards for the conduct of bio-equivalence study.
Forty subjects who met the eligibility criteria were enrolled in each of the fasting and fed study. However, only 38 subjects in fasting and 39 subjects in fed were dosed with a single dose of either test or reference products in period I. In fasting branch, two subjects, subject 26 was withdrawn from the study due to medical event and subject 37 withdrew consent due to personal reasons, both were prior to dosing of period I and in fed one subject, subject 22 was withdrawn from the study due to medical event prior to dosing of period I. Study drug was administered in sitting posture at fixed time in each period and were instructed to maintain sitting posture for 02 hours post dose. A total of 08 subjects in fasting study and 03 subjects in fed study were dropped out of the studies for different reasons.
Washout period of 02 days were maintained between each period, in order to minimize any possibility of carryover effect from preceding treatment. The blood samples were collected at pre-defined time intervals for the measurement of concentration and pharmacokinetic parameters of Dasatinib in both the periods.
Thus the data obtained from 32 subjects in fasting and 37 subjects in fed who completed the study were used for the pharmacokinetic and statistical analysis of Dasatinib.
Bioequivalence was determined by statistical comparison of Ln-transformed data of Cmax and AUC0-t of the test and reference formulations using SAS version 9.4.
Subjects drug administration and blood sampling: After an overnight fasting of 10 hours, in fasting condition subjects were administered with a single oral dose of either test product or reference product with 240 mL of water according to the sequence assigned to each subject, in sitting posture in each period.
In case of fed study all the subjects were provided with high fat high calorie breakfast 30 minutes prior to dosing, then administered with investigational product with 240 mL of water according to the sequence assigned by the randomization process.
Compliance to drug administration was assessed by examination of the oral cavity and hands of the subject immediately after dosing.
All the subjects remained in sitting posture for 02 hours after dosing. During this restriction period, the subjects were permitted to walk for reasons such as but not limited to the following: natural exigencies. Subjects were restricted from consumption of water for 01 hour before and 01 hour after dosing in each period and allowed to drink water ad libitum thereafter.
The pharmacokinetic profile of both test and reference products (in terms of rate and extent of absorption) were evaluated based on measured concentration of drug in the human plasma samples collected during the clinical phase. Blood samples for pharmacokinetic analysis were designed appropriately for characterizing the pharmacokinetic profile for the given treatments at the dose administered.
Blood samples, of 04 mL each one, were collected at 00.00 (Pre-dose), 00.17, 00.33, 00.50, 00.75, 01.00, 01.33, 01.67, 02.00, 02.33, 02.67, 03.00, 03.50, 04.00, 05.00, 06.00, 08.00, 12.00, 16.00, 24.00 and 34.00 hours post dose for measurement of pharmacokinetic parameters in fasting condition.
Blood samples, of 04 mL each one, were collected at 00.00 (Pre-dose), 00.33, 00.67, 01.00, 01.33, 01.67, 02.00, 02.33, 02.67, 03.00, 03.50, 04.00, 05.00, 06.00, 08.00, 12.00, 16.00, 24.00 and 34.00 hours post dose for measurement of pharmacokinetic parameters in fed condition.
Tolerability: Subjects were monitored for adverse events during all four periods of the study. Subjects were instructed to inform clinical personnel of any untoward medical symptoms and/or events that arose during the study. The Principal Investigator/sub-investigator/ physician of the study also evaluated the subjects for subsequent dosing. Each adverse event reported by the subjects during the study was assessed for its seriousness, severity, relationship with the study drug and outcome.
Subjects’ Safety was assessed via continuous monitoring and scheduled recording of safety measurements throughout the study through clinical examinations, vital signs assessment, 12- lead Electrocardiogram (ECG), clinical laboratory parameters (e.g. Haematology, Biochemistry, Urine analysis and Serology test) and monitoring subjects’ well-being, symptoms and signs for adverse events.
No serious adverse events were reported during the conduct of this study in fasting condition. A total of 73 adverse events were reported during the study. Thirty-five adverse events were reported following administration of reference product and thirty-eight with test product. One medical event (elevated blood pressure) has reported prior to dosing. The incidence of adverse events in the study for reference product was (52.24%) and test product was (54.29%).
No serious adverse events were reported during the conduct of this study in fed condition.
A total of 71 adverse events were reported during the study. Thirty-three adverse events were reported following administration of reference product, thirty-seven with test product and 01 during post study safety evaluation. One medical event (elevated blood pressure) has reported prior to dosing. The incidence of adverse events in the study for reference product was (43.42%) and test product was (48.68%).
All the adverse event reported in the study were resolved completely without sequelae and were assessed to be either mild or moderate in severity by the investigator/physician.
Pharmacokinetic and Statistical Analysis: Pharmacokinetic and statistical analysis was performed using the concentration data obtained from 32 subjects in fasting and 37 subjects in fed conditions who completed all four periods of the study. In order to test the two one-sided tests for bioequivalence, ratio analysis, 90% confidence intervals for the difference between treatments’ least-square mean was calculated for Ln-transformed Cmax and AUC0-t of Dasatinib.
Pharmacokinetic parameters were calculated using noncompartmental model of Phoneix® WinNolin® version 8.1 and statistical analysis was carried out using the SAS® statistical software, version 9.4 of SAS Institute Inc, USA.
The mean, standard deviation, standard error, geometric mean, coefficient of variation, minimum, median, maximum and range were calculated for Cmax, AUC0-t and Tmax.
RESULTS AND DISCUSSION
Forty subjects participated in the study and thirty-two subjects in fasting and thirty-seven subjects in fed completed all four periods of the study. The clinical study was conducted over a period of 10 days (fasting) and 09 days (fed). Blood sampling was done at pre-defined intervals up to 34.00 hours in all four periods, separated by a washout period of 02 days.
Though the number of adverse events reported during the study is high in number, these are comparable with the previous data available in published studies. Hence, the test and reference products can be considered well tolerated in healthy subjects at a given dose. All the adverse events reported are mild to moderate in intensity and resolved either during the course of the study without sequelae.
The pharmacokinetic parameters observed in the study for both the test and reference products are similar to that of the values reported in previous published data.
The pharmacokinetic plasma samples collected from the subjects in both conditions were analysed to determine concentration of Dasatinib using a validated bio-analytical method in LCMS/MS. The pharmacokinetic and statistical analyses of Dasatinib were performed using the concentration data obtained from subjects who completed all four periods of the study.
Fasting Condition: The ISCV of Cmax for reference product was calculated to be 104.94%, hence, 90% confidence interval limit was widened using scaled-average-bioequivalence and the value obtained 72.44% - 123.72% was found to be within the widened limits of 69.84% to 143.19%.
| Table 1: Statistical Results of Test Product-T versus Reference Product - R for Dasatinib (Fasting) (N=32) | |||||||
| Parameters | Antilog Least Square Mean | Point Estimate (%) | 90%Confidence Interval | ISCV (%)R vs R | ISCV (%)T vs R | Power | |
| Test Product (T) | Reference Product (R) | ||||||
| Ln (Cmax ) | 151.6071 | 160.1441 | 94.67 | 72.44% - 123.72% | 104.94 | 110.8 | 37.87 |
| Ln (AUC0-t) | 691.6659 | 694.4015 | 99.61 | 84.84% - 116.94% | 61.55 | 57.76 | 72.79 |
| Table 2: Statistical Results of Test Product-T versus Reference Product - R for Dasatinib (Fed) (N=37). | ||||||||
| Parameters | Antilog Least Square Mean | Point Estimate (%) | 90% Confidence Interval | ISCV (%) R vs R | ISCV (%) T vs R) | Power (%) | ||
| Test Product (T) | Reference Product (R) | |||||||
| Ln (Cmax ) | 107.6152 | 96.895 | 111.06 | 102.34% - 120.53% | 30.92 | 30.68 | 99.62 | |
| Ln (AUC0-t) | 593.8994 | 557.1844 | 106.59 | 100.72% - 112.80% | 26.38 | 20.97 | 100 | |
| Table 3: Summarized Demographic Profile of Subjects who Completed the Study for Dasatinib (Fasting) (N=32). | ||||
| Parameter | Mean | SD | Minimum | Maximum |
| Age (years) | 31 | 6 | 22 | 43 |
| Height (m) | 1.699 | 0.058 | 1.583 | 1.836 |
| Weight (Kg) | 71 | 8.2 | 50 | 88.9 |
| BMI (Kg/m2) | 24.58 | 2.5 | 18.5 | 28.87 |
| Table 4: Summarized Demographic Profile of Subjects who Completed the Study for Dasatinib (Fed) (N=37). | ||||
| Parameter | Mean | SD | Minimum | Maximum |
| Age (years) | 29 | 4 | 22 | 41 |
| Height (m) | 1.706 | 0.066 | 1.54 | 1.83 |
| Weight (Kg) | 69.9 | 9.8 | 54.8 | 88.8 |
| BMI (Kg/m2) | 23.99 | 2.83 | 18.75 | 28.1 |
| Table 5: Summary of Pharmacokinetic Parameters for Dasatinib (Fasting) of Reference Product -R. | ||
| Parameter | N | Reference (R) (Mean ± SD) |
| Cmax (ng/mL) | 64 | 203.852 ± 95.458 |
| AUC0-t (ng.hr/mL) | 64 | 793.540 ± 313.142 |
| *Tmax (hr) | 64 | 1.00 (0.50 - 24.00) |
| *Expressed in terms of median (range) | ||
| Table 6: Summary of Pharmacokinetic Parameters for Dasatinib (Fasting) of Test Product -T. | ||
| Parameter | N | Test (T) (Mean ± SD) |
| Cmax (ng /mL) | 64 | 211.698 ± 107.329 |
| AUC0-t (ng.hr/mL) | 64 | 820.124 ± 347.389 |
| *Tmax (hr) | 64 | 1.00 (0.50 - 24.00) |
| *Expressed in terms of median (range) | ||
| Table 7: Summary of Pharmacokinetic Parameters for Dasatinib (Fed) of Reference Product -R. | ||
| Parameter | N | Reference (R) (Mean ± SD) |
| Cmax (ng/mL) | 74 | 104.437 ± 36.788 |
| AUC0-t (ng.hr/mL) | 74 | 584.312 ± 172.250 |
| *Tmax (hr) | 74 | 2.00 (1.00 - 6.00) |
| *Expressed in terms of median (range) | ||
| Table 8: Summary of Pharmacokinetic Parameters for Dasatinib (Fed) of Test Product –T. | ||
| Parameter | N | Test (T) (Mean ± SD) |
| Cmax (ng /mL) | 74 | 118.674 ± 51.814 |
| AUC0-t (ng.hr/mL) | 74 | 622.537 ± 190.525 |
| *Tmax (hr) | 74 | 2.00 (0.67 - 6.00) |
| *Expressed in terms of median (range) | ||
90% confidence interval of AUC0-t was 84.84% - 116.94%, which was within the acceptable limits of 80.00% to 125.00%.
Fed Condition: The ISCV of Cmax for reference product was calculated to be 30.92%. 90% confidence interval of Cmax and AUC0-t were 102.34% - 120.53% and 100.72% - 112.80% respectively, which were found to be within the acceptable Limits.table (9,10)
| Table 9: p -Value for Cmax and AUC of Dasatinib (Fasting). | |||
| Parameters | Cmax | AUC0-t | Significance |
| Sequence effect | 0.062 | 0.0714 | Significant for Cmax and AUC0-t |
| Period effect | 0.4654 | 0.378 | Insignificant for Cmax and AUC0-t |
| Treatment (Formulation) effect | 0.7345 | 0.9675 | Insignificant for Cmax and AUC0-t |
| P < .05 for Period and Treatment (Formulation) effects, P < .10 for Sequence effect | |||
| Table 10: p -Value for Cmax and AUC of Dasatinib (Fed) | ||||
| Parameters | Cmax | AUC0-t | Significance | |
| Sequence effect | 0.4882 | 0.4489 | Insignificant for Cmax and AUC0-t | |
| Period effect | 0.2136 | 0.3 | Insignificant for Cmax and AUC0-t | |
| Treatment (Formulation) effect | 0.0357 | 0.0642 | Significant for Cmax and Insignificant for AUC0-t | |
| P < .05 for Period and Treatment (Formulation) effects, P < .10 for Sequence effect | ||||
CONCLUSION
The study is aimed to evaluate the bioequivalence between the test product of Abbott, Colombia vs the marketed product, Sprycel of Bristol- Myers in healthy subjects in both fasting and fed conditions.
The bioavailability of test and reference products was compared after administering the same to healthy subjects.
Bioequivalence was demonstrated between Dasatinib 100 mg film-coated tablets of Abbott Laboratories versus Sprycel® (Dasatinib) 100 mg film-coated tablets of Bristol-Myers Squibb under both fasting and fed condition in healthy subjects. Both the test and reference products were relatively well tolerated at the selected dose levels by the selected study population.
REFERENCES
1. International Conference on Harmonization (ICH): Harmonized Tripartite Guideline– Guideline for Good Clinical Practice (GCP) - E6, 2016.
2. Structure and Content of Clinical Study Reports E3 Current Step 4 version dated 30 November 1995.
3. CDSCO’s New Drugs and Clinical Trials Rules 2019 G.S.R. 227(E).
4. 21 Code of Federal Regulations.
5. Prescribing information of Sprycel® Bristol-Myers Squibb Pharma USA updated on 21- DEC-2018.
6. EMA guideline on the investigation of bioequivalence, 2010.









































































































































{kind=link}