3-Hydroxypropanal (Reuterin) the Overlooked Cyclophosphamide Metabolite
- 1. Institute of Biochemistry II. Goethe University Frankfurt Medical School, Frankfurt Germany
CITATION
Voelcker G (2022) 3-Hydroxypropanal (Reuterin) the Overlooked Cyclophosphamide Metabolite. JSM Biochem Mol Biol 8(1): 1038.
INTRODUCTION
Cyclophosphamide (CP) is one of the oldest anticancer drugs, but one that is also highly effective by today’s standards. The theoretical idea for the development of CP was to administer a nontoxic prodrug that liberates in tumor tissue nitrogen mustard which damages DNA by alkylation. At the time, when CP was de veloped, it was believed that enzymes that catalyze splitting of phosphorus–nitrogen bonds (phosphoamidases) were preferen tially found in tumor tissue [1,2]. This was the basis for the de velopment of pro drugs from which alkylating nitrogen mustard should be split off in the target tissue “tumor”. Of the substances synthesized based on this theoretical requirement, CP proved to be the most effective in animal experiments. The theoretical concept was wrong, but the result was a very effective anticancer drug.
Brief history of the metabolism of CP
Very soon it turned out that CP is completely ineffective against tumor cells in vitro. Substances that kill tumor cells in vit ro are only detectable in the blood after injection of CP. From the stable but therapeutically ineffective CP metabolites 4-ketocyclo phosphamide (keto-CP) and carboxyphosphamide (CARB) found in the blood after CP injection, it was concluded that CP is hydrox ylated by liver cytochrome P450 enzymes to form 4-hydroxycy clohosphamide (CPOH). which forms an equilibrium with its tau tomer aldophosphamide (ALD) [3]. In vitro experiments in phos phate buffer solution with synthetically prepared CPOH showed that the alkylating agent was phosphoramide mustard (PAM) formed by β-elimination of acrolein from ALD [4]. PAM was as sumed to kill cells by DNA alkylation. The detection of CPOH (Fig ure 1) after CP (Figure 1) injection was initially difficult because no reference substance was available.
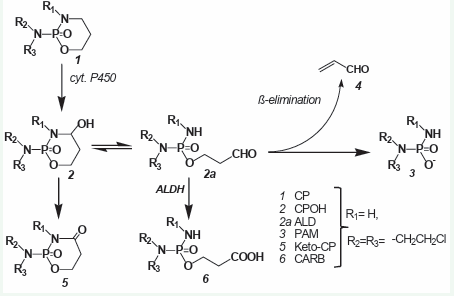
Figure 1: Historical overview of the metabolism of CP..
Only after a synthesis for CPOH was developed [5], it was possible to prove CPOH in blood by thin layer chromatography and mass spectrometry [6,7], after CP injection. This was the state of knowledge about the metabo lism of CP that had been frozen for decades and on which hypoth eses about the mechanism of action and possibilities for improv ing the therapeutic effectiveness of CP were based.
The two reactions competing for ALD, detoxification to CARB by aldehyde dehydrogenases (ALDH) and formation of PAM from ALD by β-elimination of acrolein have long been the focus of in terest because they explain the different sensitivity of different tissues to CP. Cells with high ALDH activity such as intestinal mu cosal cells, hematopoietic stem cells or occult tumor cells in bone marrow transplants [8], that can be eradicated with congeners of CP such as Mafosfamide (MF), which spontaneously hydrolyses to CPOH have low activity of ALDH. But attempts to explain there by the tumor specificity of CP [8], were not convincing because by co-treatment with inhibitors of ALDH CP resistant tumors did not become CP sensible.
Historical overview of the metabolism of CP
After application of CP [1], in animals and patients, the stable but therapeutically ineffective metabolites ketocyclophospha mide [2], and CARB [6], were detected in the blood and urine. It was concluded that CP is hydroxylated by liver cytochrome P450 enzymes to give CPOH [2], which is in equilibrium with its tautomer ALD (2a). In vitro experiments with synthetic CPOH in buffer solutions showed that the alkylating agent PAM [3], is formed from ALD by ß-elimination of acrolein [4].
After intravenous injection of 100 mg/kg CP in mice, 92% is hydroxylated to CPOH. 80% of this is detoxified to Keto-CP and CARB [9].
The old metabolite scheme of CP solidified over time because it was the basis for the very successful introduction of mercap toethanesulfonic acid (Mesna) as an antidote to CP -and Ifosfa mide (IF) toxicity. According to the old notion of CP metabolism, acrolein was the main cause of CP and IF dose limiting hemor rhagic cystitis, which is successfully treated with 2-mercap toethanesulphonate (MESNA), It was believed that Mesna exert their toxicity-reducing effect by forming inert thioethers with acrolein [10].
Studies of the breakdown of CPOH in rat serum and protein free rat serum ultrafiltrate demonstrated that CPOH breaks down more than 100 times faster in rat serum than in rat serum ul trafiltrate. Phosphodiesterases (PDE) of the snake venom phos phodiesterase type (EC 3.1.4.1) were identified as the enzymes responsible for this decomposition. [11,12]. This was nothing exciting per se, but the whole thing became exciting when the reaction products of the decomposition of the tautomers CPOH/ ALDO were identified. The reaction products are not acrolein and PAM but PAM and 3-hydroxypropanal (HPA) [13]. However, at the time HPA was discovered as a CP metabolite it was assumed that HPA was only an insignificant intermediate product on the metabolic pathway to PAM, the discovery was not taken serious ly. This is probably also why HPA did not fit into the worldview in which acrolein is the cause of hemorrhagic cystitis. The real cause of heorrhagic cystitis after CP and IF application is CPOH (IFOH) itself, which reacts with nucleophilic groups, e.g., SH groups of membrane proteins and is neutralized by Mesna [14].
The real metabolism of CP
PAM [3] is formed from ALD (2a) by enzymatic cleavage of ALD by phosphodiesterases (PDE). This creates HPA [7].
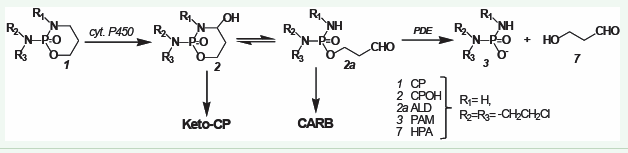
Figure 2: The real metabolism of CP..
Implications of the discovery of HPA as a CP metabo lite for CP’s mechanism of action
After the discovery of apoptosis, the view of the mechanism of action of alkylating substances changed. It has become apparent that any substance that damages DNA thereby initiates apoptosis.
Schwartz and Waxman [15], investigated the effect of MF on the caspase 8 (extrinsic) and caspase 9 (intrinsic) depend ent pathways of apoptosis in 9L tumor cells and 9L tumor cells transduced with CP hydroxylating CYP2B6. Contrary to other an ticancer drugs like doxorubicin [16], and cisplatin [17], in which activation of caspase 8 is the initial apoptotic event, after appli cation of MF, activation of caspase 9 was detectable before the activation of caspase 8. In addition caspase 9 was activated to a greater extent than caspase 8, indicating the p53 mediated apop totic pathway, in response to upstream DNA damage, is triggered by CPOH released in minutes from MF. Seki et al came to the same conclusion, showing that. caspase 8 specific inhibitors only block cisplatin but not CP induced [16].
In the functional mechanism of alkylating agents, Schwartz and Waxman [15], distinguish between a cytostatic and a cyto toxic effect. In 9L cells, overexpressing the anti-apoptotic Bcl-2 protein, they could only determine the cytostatic effect after treatment with MF but no cytotoxic apoptosis. They state that the combination of CPOH induced DNA damage and Bcl-2 depend ent cytotoxic response is necessary for cell death. The mentioned reports clearly demonstrate that the functional mechanism of OHCP is bimodal: First occurs the cytostatic DNA damage by PAM and, secondly, the cytotoxic p53 mediated apoptosis is observed. The second step is critical because it can be manipulated by fac tors that affect the pathway of apoptosis. Such a factor is very likely HPA, one of the two reaction products of the enzymatic de composition of OHCP.
HPA, also known as reuterin, is produced by Lactobacillus re uteri and submitted to the culture medium. It has been shown to be active against bacteria viruses and fungi [18], it is used as food additive to prevent spoilage by growth of pathogens. Experiments by Iyer [19], who investigated the effects of the supernatant of L. reuteri cultures (LR) on tumor necrosis factor (TNF)-activated apoptosis signaling pathways in human leukemia cells, showed that reuterin inhibits (i) the formation of the anti-apoptotic pro teins Bcl-2 and Bcl-xL and (ii) the TNF dependent NF-κB activa tion by inhibition of the translocation of the p65 subunit of NF-κB into the nucleus. The experiments of Iyer et al. further showed that the degradation of IκBα by lack of the ubiquitination of IκBα is suppressed by the supernatant of LR. Thus, by the inhibition of the nuclear translocation of NF-κB apoptosis is enhanced [19].
In summary, the scientific literature gives the following picture:
1. The event leading to cell death after CP application is not DNA damage but apoptosis triggered by DNA damage [5].
2. The CP metabolite HPA is an apoptosis enhancer. This leads to the action scheme of CP and other OX shown in (Figure 3).
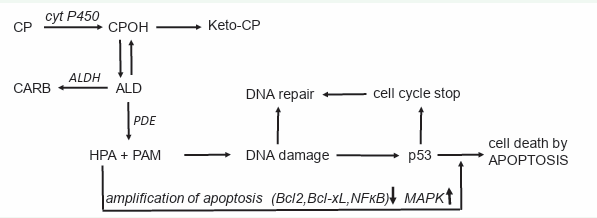
Figure 3: Mechanism of action of CP and other OX.
Mechanism of action of CP and other OX
CP (and other OX) is hydroxylated by P450 enzymes in the liv er to CPOH from which a small part is oxidized to keto-CP. CPOH is in equilibrium with its tautomere ALD. ALD is the pharmaco logic active metabolite. The bulk of it is oxidized by ALDH to the nontoxic CARB. The amount of ALD not oxidized, is decomposed by phosphodiesterase (PDE) to the alkylating PAM and proapop totic HPA. PAM damages DNA by alkylation. The alkylated DNA is either repaired immediately or, if this is not possible, the tumor suppressor protein p53 is activated, which induces cell cycle stop to give the cell time to repair the damage. If DNA repair is not possible, p53 induces apoptosis, which is - and this is special for CP and other OX - enhanced by HPA which supports apoptosis by inhibiting the anti-apoptotic proteins Bcl-2 and Bcl-xL, and by inhibiting NF-κB activation. Additional HPA promotes apoptosis by enhancing mitogen-activated protein kinase (MAPK) activities by enhancing JNK and p38 phosphorylations.
EXPERIMENTAL EVIDENCE FOR PRESENTED ME CHANISM OF ACTION OF CP AND OTHER OX
In experiments on regional perfusion of the tumor-bearing limb in rats, CPOH proved to be completely ineffective in contrast to systemic therapy with CP [20, 21]. This was despite the fact that the CPOH concentration in the perfusate (oxygenated hemo globin solution) at the point of entry into the rat leg (a. epigastric superficialis) was about 100 times higher than after CP applica tion. The amount of CPOH taken up by the tissue was 12 times greater in the perfusion experiments than after systemic therapy with CP [22].
In perfusion experiments with nitrogen mustard N-oxide, which has a comparable effect to PAM, and in experiments with cisplatin, the animals were cured. The reason for the failure of the perfusion experiments with CPOH was clearly that the perfusion solution did not contain ALD-splitting PDE.
CP was “found” by a happy coincidence. It is not tailor-made for the reaction scheme in Figure 3. Disadvantages are the indi vidually varying hydroxylation by cyt. p450 enzymes in the liver, the formation of toxic CPOH en route to the pharmacologically active metabolite ALD, and the lack of adaptation of the alkylat ing function for DNA damage that initiates optimal apoptosis ex ploitation. To correct the first two CP “design errors” mentioned, tiazolidine and perhydrothiazines were synthesized from aldo phosphamide and I-aldophosphamide, which hydrolyze sponta neously, bypassing toxic CPOH (IFOH) to ALD (IALD) [23] (Figure 4).
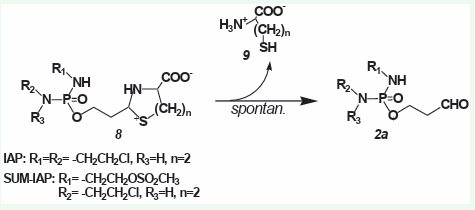
Figure 4: Spontaneous hydrolysis of thiazolidines (8 n=1) and perhydrothiazines (8 n=2) from aldophosphamide (8 R1 =H, R2 =R3 = -CH2 CH2 Cl) and I-aldophosphamide (8 R1 =R2 = -CH2 CH2 Cl, R3 =H) to ALD (2a) and L-cys (9 n=1) or homocysteine (9 n=2).
Spontaneous hydrolysis of thiazolidines (8 n=1) and perhy drothiazines (8 n=2) to ALD (2a, R1 =H, R2 =R3 = -CH2 CH2 Cl) and I-aldophosphamide (2a, R1 =R2 = -CH2 CH2 Cl, R3 =H) and L-cys (9 n=1) or homocysteine (9 n=2).
These compounds are nine fold lower toxic than CPOH and more effective than the parent compounds CP and IF against subcutaneously transplanted P388 tumors in mice [23]. A spe cial feature of the thiazolidine compounds is that the thiazolidine ring gives them the ability to cross the blood-brain barrier [24]. Because the perhydrothiazine of I-aldophosphamide (Figure 4), R1 =R2 = -CH2 CH2 Cl, R3 =H, n=2) is easy to synthesize and easy to handle, further experiments with this compound (I-aldophos phamide perhydrothiazine, IAP ) were carried out.
Another construction flaw of CP - the lack of adaptation to the initiation of apoptosis - can be corrected as follows: Accord ing to the scheme (Fig.3), the apoptosis yield is greater, the lower the amount of alkylated DNA that is repaired by the cell’s own repair mechanisms. According to Povirk and Shuker [25], chlore thyl groups as found in IAP form interstrand cross links. whereas methane sulfonate groups form intrastrand cross-links [22]. In contrast to interstrand cross-links, intrastrand cross-links are poorly repaired (http://www.atdbio.com/content/16/Nucleic-acid-drug interactions).
If in the IAP molecule (Figure 4) in the residue R1 (-CH2 CH 2 Cl) the chlorine is substituted by a sulfonyl methyl group (R1 is now -CH2 CH2 OSO2 CH3 ), a perhydrothiazine derivative called SUM-IAP is obtained. SUM-IAP is in cell culture experiments with P388 mice leukemia cells significantly less cytotoxic than IAP but against the same cells when growing as solid tumors in mice 104 105 times more effective than IAP [26].
These experimental results - the failure of CPOH in regional perfusion of the tumor-bearing limb in rats and the enormous increase in antitumor effect when repair of the alkylated DNA is prevented - are a strong indication that the scheme in (Figure 3) is correct.
One can discuss whether HPA, the long-neglected CP metabo lite, makes CP, long classified as an alkylating cytostatic, to a new class of cytostatics, the class of apoptosis boosters.









































































































































{kind=link}