Are the SARS-Cov-2 Variants Greater Threats?
- 1. Department of Chemistry, Drexel University, USA
Abstract
Two years have passed since the beginning of the SARS-CoV-2 pandemic and the continual emergence of SARS-CoV-2 mutated viral strains has led to continual re-evaluation of the current therapeutic options. Herein we continued an analysis of the interactions of SARS-CoV-2 Spike (S), protein with human angiotensin converting enzyme (ACE-2), and compared these interactions with those of the variants of concern (VOCs), and variants of interest (VOIs), that have emerged since August 2021 with ACE-2. We have also compared the binding affinities of several possible S-protein inhibitors with the binding affinity of these same inhibitors with the VOCs and VOIs by means of in silico molecular docking studies. The highest binding potential inhibitor was complexed with the spike proteins and docked with ACE-2. The protein-protein binding decreases when the inhibitor is bound to the S-proteins, however the binding affinity to the inhibitors varies. The inhibitors show decreased binding affinity to the VOCs mutated S-proteins and similar affinity to the VOIs in comparison to SARS-CoV-2, indicating the variability of binding amongst the variants deems the S-protein a poor target for the therapeutic approach.
Keywords
SARS-Cov-2; In silico molecular docking; Angiotensin converting enzyme (ACE-2).
CITATION
Wilson J, Hai-Feng J (2022) Are the SARS-Cov-2 Variants Greater Threats? – A Continued in silico Analysis of the Spike Protein. JSM Biochem Mol Biol 8(1): 1037.
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS CoV-2), which causes the coronavirus disease (COVID-19), is a beta coronavirus first reported in December 2019 in Wuhan, China [1]. Initiating a global pandemic, SARS-CoV-2 has caused over 300 million confirmed cases of COVID-19 and over 5 million reported deaths [2]. Since our first reported work [3], four new mutated strains have emerged, Lambda, Mu, Omicron BA.1, and Omicron BA.2, with the latter two being designated as variants of concern (VOCs), by the World Health Organization [4]. Omicron sub-variants BA.1 and BA.2 have caused higher infection rates and brought questions of continued therapeutic efficacy to the forefront [5,6]. The Lambda and Mu variants, designated as variants of interest, are no longer circulating.
As viral entry is facilitated by the spike protein (S-protein), it is an important protein target for therapeutic routes [7,8]. The S-protein consists of 1260 amino acids. It is composed of 2 sub units: S1 (672 amino acids), and S2 (588 amino acids) [9,10]. S1 contains the receptor binding domain and binds with the human angiotensin-converting enzyme (ACE-2), receptor on host cells. The S2 subunit facilitates fusion of the membrane [11,12].
All the variant strains have mutations at the receptor binding domain, not only increasing VOC infectivity but also raising the question of continued therapeutic efficacy [13,14]. In this study we calculate and compare the binding energies of ACE-2 with the S-proteins of SARS-CoV-2 and the four recent variants. In addition, we docked the previously reported top ten potential S-protein-ACE2 inhibitors from a library of FDA approved drugs with the new variants and compared the binding affinities to the original S-protein. The highest binding potential inhibitor was then complexed to the S-proteins and docked again with ACE-2. As mentioned, this study is a continuation of our earlier reported work on in silico modeling of the SARS-CoV-2 S-protein and 3CL Protease [3,15,16].
METHODS
Molecular docking calculations were performed using AutoDock Vina 1.1.2 [17], Ligands were prepared using AutoDockTools-1.5.6 [18], Chimera 1.14 [19], and Avogadro [20]. Proteins were mutated in silico using Schrödinger Maestro [21] and prepared using AutoDockTools-1.5.6. Protein-ligand complexes were visualized with PyMOL [22] and ChimeraX-1.2.1 [23]. Protein-protein docking was performed utilizing the HADDOCK webserver [24].
Preparation of Receptor and Ligands
The crystal structure of the SARS-CoV-2 S protein RBD bound to ACE-2 was retrieved from the Protein Data Bank (PDB ID: 6M0J). Using AutoDockTools the structure was cleaned, removing ACE-2, and prepared through the addition of polar hydrogens and calculation of Gasteiger charges. A databank of FDA-approved drugs was retrieved from BindingDB to be used as ligands in the docking study. The library of ligands were prepared and optimized with the MMFF94 force field in Avogadro.
In silico Mutation of Spike Protein
The SARS-CoV-2 S protein RBD was mutated in silico and then prepared in the same method as described above for the receptor before docking. Using Shrödinger Maestro, individual amino acid residues were selected and altered to match the mutations of the variants. For Omicron BA.1 the following mutations were carried out: G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493 R, G496S, Q498R, N501Y, and Y505H. For Omicron BA.2: G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, and Y505H. Two amino acids were mutated for the Lambda variant: L542Q, and F490S. And for the Mu variant: R346K, E484K, and N501Y [25].
Molecular Docking
Spanning 22.00 Å × 42.00 Å × 22.00 Å along the x, y and z axis, respectively, and centered at x = -27.878, y = 25.205 and z = 5.514 the grid box was constructed to cover the SARS-CoV-2 ACE-2 binding region to ensure inhibition of the interaction. The 4000+ FDA-approved drug ligand library was docked with the S-protein and the top ten highest binding ligands by docking score (kcal/ mol), were selected. The highest binding affinity being the most negative value. The top ten ligands were then docked with the mutated S-proteins to determine how the mutations would affect the binding affinity.
Visualization with PyMOL and Chimera
The protein-ligand complex of the highest binding affinity for the S-protein was analyzed using both PyMOL and Chimera to visualize the ligand conformation and interactions between the protein and ligand. The complex between this same ligand and the mutated proteins were also visualized, regardless of docking score to compare ligand conformation and interactions.
Protein-Protein Docking
The HADDOCK webserver was utilized to dock the S-protein and mutated S-proteins with human ACE-2. And then again to dock the S-proteins- Irinotecan complexes to ACE-2. Protein ligand complexes were constructed using ChimeraX. Active residues were input based on the work of Lim et al. [26]. Results are reported as a HADDOCK score, a unitless weighted sum of energy terms.
RESULTS
The resulting protein-protein docking scores between the S-protein RBDs and ACE-2 are tabulated in Table 1.
| Table 1: HADDOCK Score of ACE-2 with SARS-CoV-2 S-protein or the S-protein variants. | ||
| S-protein with ACE-2 | HADDOCK score | p-value |
| SARS-CoV-2 S-protein | -134.3 +/- 3.4 | |
| SARS-CoV-2 S-protein Omicron BA.1 | -142.5 +/- 6.5 | 0.38 |
| SARS-CoV-2 S-protein Omicron BA.2 | -146.1 +/- 4.5 | 0.53 |
| SARS-CoV-2 S-protein Lambda | -137.1 +/- 1.5 | 0.59 |
| SARS-CoV-2 S-protein Mu | -136.3 +/- 1.8 | 0.65 |
The HADDOCK scores of the variant S-protein-ACE2 binding have been compared to the original S-protein score. Table 2 compiles the top ten highest binding affinities of the FDA-approved drug library with the SARS-CoV-2 S-protein RBD compared to the affinities of the same drugs bound to the RBD variants.
| Table 2: Binding affinity of highest binding FDA ligands and SARS-CoV-2 S-protein RBD compared to same ligands docked with VOC and VOI spike proteins. | ||||||
| Drug Name | Binding Affinity (kcal/mol) | Indication | ||||
| Original | Omicron BA.1 | Omicron BA.2 | Lambda | Mu | ||
| Irinotecan | -8.6 | -7.6 | -7.5 | -8.7 | -8.4 | Metastatic Carcinoma |
| CAS # 2415492-58-7 | -8.5 | -7.0 | -7.0 | -9.0 | -4.7 | |
| Dihydroergotamine | -8.5 | -8.0 | -8.5 | -8.5 | -7.7 | Migraine headaches |
| Lapatinib | -8.4 | -7.0 | -6.4 | -8.6 | -7.3 | Metastatic Breast Cancer |
| Nilotinib | -8.4 | -7.6 | -7.7 | -8.3 | -8.6 | Myeloid Leukemia |
| Lapatinib ditosylate | -8.4 | -7.2 | -6.9 | -7.9 | -7.2 | Metastatic Breast Cancer |
| Digoxin | -8.3 | -7.9 | -7.4 | -8.4 | -7.6 | Heart Failure |
| Digitoxin | -8.3 | -8.1 | -7.4 | -8.5 | -7.4 | Congestive heart failure |
| CAS # 1610051-53-0 | -8.3 | -7.6 | -7.3 | -8.6 | -8.9 | |
| Ergotamine | -8.3 | -7.9 | -7.7 | -8.5 | -8.5 | Migraine headaches |
| p-value | 0.0001 | 0.0002 | 0.31 | 0.07 | ||
The docking scores are arranged in decreasing affinity with respect to the S-protein RBD. The highest binding ligand, irinotecan, a chemotherapy drug, was complexed with the S-proteins and docked with ACE2 to demonstrate the inhibition potential. Evaluation of these HADDOCK scores are in Table 3 and have been compared to the scores in Table 1.
| Table 3: HADDOCK score of S-protein-irinotecan complex docked with ACE-2. | |||
| S-protein with ACE-2 |
HADDOCK score S-protein + ACE2 |
HADDOCK score S-protein + Inhibitor + ACE2 |
p-value |
| SARS-CoV-2 S-protein | -134.3 +/- 3.4 | -113.5 +/- 2.0 | |
| SARS-CoV-2 S-protein Omicron BA.1 | -142.5 +/- 6.5 | -103.8 +/- 5.3 | 0.38 |
| SARS-CoV-2 S-protein Omicron BA.2 | -146.1 +/- 4.5 | -87.6 +/- 4.9 | 0.53 |
| SARS-CoV-2 S-protein Lambda | -137.1 +/- 1.5 | -112.0 +/- 6.4 | 0.59 |
| SARS-CoV-2 S-protein Mu | -136.3 +/- 1.8 | -143.1 +/- 3.2 | 0.65 |
Figure 1 depicts the docking of the highest binding ligand, irinotecan, to the S-protein RBD, as well as the mutated S-proteins.
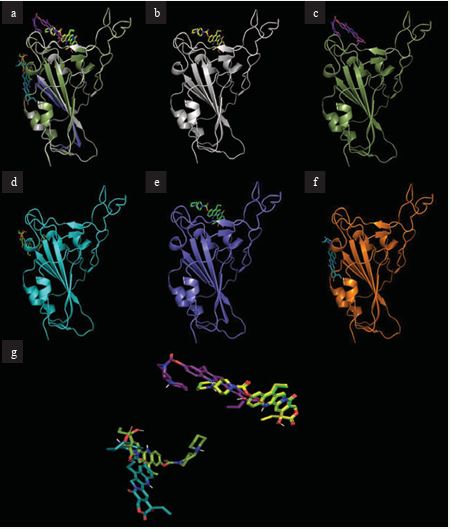
Figure 1: Docking site of irinotecan with S-proteins RBDs. (a) Overlay of all S-proteins bound to irinotecan (b) Irinotecan bound to S-protein. (c) Irinotecan bound to S-protein RBD Omicron BA.1. (d) Irinotecan bound to S-protein RBD Omicron BA.2. (e) Irinotecan bound to S-protein RBD Lambda. (f) Irinotecan bound to S-protein RBD Mu. (g) Overlapping structures of irinotecan in bound conformation to all protein RBDs.
Figure 2 illustrates the interactions of this binding event.
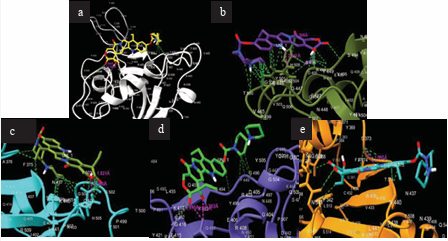
Figure 2: Interactions between irinotecan and SARS-CoV-2 Proteins. (a) Irinotecan bound to S-protein. (b) Irinotecan bound to S-protein RBD Omicron BA.1. (c) Irinotecan bound to S-protein RBD Omicron BA.2. (d) Irinotecan bound to S-protein Lambda. (e) Irinotecan bound to S-protein RBD Mu.
Figure 3 shows the docking conformation of the S-protein-irinotecan-ACE2 system.
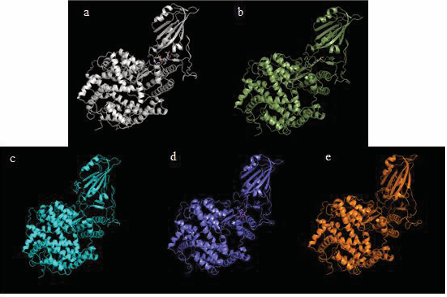
Figure 3: S-protein(s), irinotecan and ACE-2 docked. (a) Irinotecan bound to S-protein docked with ACE-2. (b) Irinotecan bound to S-protein RBD Omicron BA.1 docked with ACE-2. (c) Irinotecan bound to S-protein RBD Omicron BA.2 docked with ACE-2. (d) Irinotecan bound to S-protein Lambda. (e) Irinotecan bound to S-protein RBD Mu docked with ACE-2.
DISCUSSION
The binding affinities of all the variants to ACE-2 are greater than that of SARS-CoV-2, yet as evidenced by p-values > 0.05, the difference is not statistically significant. However, the binding affinities of the S-proteins to ACE-2 serve as a comparison to the binding of the S-protein-irinotecan complex to ACE2. Here (Table 3), we do see a binding inhibition in four of the S-proteins; the original, Omicron sub-variants BA.1 and BA.2, and Mu. The identified potential inhibitor, irinotecan, does cause a weakened binding between the S-protein and ACE-2. It does not inhibit the protein-protein interaction for the Mu variant, most likely due to the binding location of irinotecan.
The decreasing binding affinity of the top ten ligands from the FDA-approved drug library with the VOC S-proteins demonstrates the therapeutic difficulties faced when targeting the spike protein. The binding affinities of the Omicron variants vary significantly from the original S-protein, meaning less susceptibility to treatment. The Lambda and Mu variants, on the other hand, are similar in binding affinity and as such explains why this variant is only designated as a variant of interest by the WHO as it most likely will be susceptible to current treatments.
CONCLUSION
The continued efficacy of the SARS-CoV-2 vaccines has been a central question as mutations to the S-protein’s receptor binding domain continue. Overall, we continue to assert that the S-protein is not the best protein target for vaccinations or therapeutic strategies. While we did demonstrate the potential of an inhibitor in hindering the binding of the S-protein to ACE2, the decreased binding to possible therapeutic options in comparison to the original S-protein for the VOCs suggests the spike proteins receptor binding domain is becoming increasingly adaptable and less susceptible to current preventative and treatment options.
REFERENCES
2. World Health Organization. WHO Coronavirus (COVID-19). Dashboard. 2022.
4. World Health Organization. Tracking SARS-CoV-2 variants. 2022.
6. Prevention, C. f. D. C. a. Omicron Variant: What You Need to Know.
22. Schrödinger L. The PyMOL Molecular Graphics System, Version 2.0. 2015.









































































































































{kind=link}