Computer Based Drug Design of Various Heterocyclic Compounds having Anticancer Activity: A Brief Review
- 1. Department of Pharmaceutical Chemistry, Y.B. Chavan College of Pharmacy, India
Abstract
This review gives the general introduction of cancer and focuses on the scheme of Docking. The current study employed Molecular Docking to develop global models capable of predicting the bioactivity of various heterocycles which are used in treatment of cancer. A wide variety of chemical structures, biological activities and Docking results of various compounds acting on cancer were collected from the various database website. Several approaches are widely being used as important tools for drug discovery such as Hansch method, Free-Wilson method, Conventional 2-D/3-D QSAR methods, Molecular Modeling and Molecular Docking. Herein, we describe the application of Docking and in addition provide hints for designing new molecule with improved activity. The concept of Docking has typically been used for drug discovery and development and has gained wide applicability for correlating molecular information with not only biological activities but also with other physicochemical properties. There are a diverse group of pharmacologically active heterocyclic compounds, most of which are clinically used. This review aims to highlight docking research reported on biological potential of substituted heterocycles. This review aims to cover the essential concepts and techniques that are relevant for performing Docking studies through the use of selected examples from previously reported work.
Citation
Nikaljea AP, Bahetia K (2017) Computer Based Drug Design of Various Heterocyclic Compounds having Anticancer Activity: A Brief Review. J Bioinform, Genomics, Proteomics 2(1): 1014.
Keywords
• Cancer
• CADD
• Molecular Docking
• Heterocycles
• 2-D/3-D QSAR
ABBREVIATIONS
ADMET: Absorption; Distribution; Metabolism; Excretion and Toxicology; CADD: Computer Based Drug Design (CADD); CDKs: Cyclin-Dependent Kinases; DHFR: Dihydrofolate Reductase; DMPK: Dystrophia Myotonica Protein Kinase; EGFR: Epidermal Growth Factor Receptor; HeLa: Human cervical cancer cell lines; Hsp90: Heat Shock Protein 90; HTS: High throughput screening; HepG2: Hepatocellular/ Hepatic carcinoma cell line; LDH-5: Lactate dehydrogenase enzyme (LDH-5); MD: Molecular Docking; MM : Molecular Modeling; MOE: Molecular Operating Environment; NAT2: N-acetyltransferase 2; NCBI: National Center for Biotechnology Information; PTEN: The Phosphatase and tensin homolog located on chromosome 10q23 (PTEN); PDB: Protein Data Bank; QSAR: Quantitative Structure-Activity Relationship; RMSD: Root Mean Square Deviation; SARs: Structure Activity Relationships; SBDD: Structure Based Drug Design; 3D:Three dimensional; TQ: Thymoquinone
INTRODUCTION
Cancer is a disease characterized by uncontrollable, irreversible, independent, autonomous, uncoordinated and relatively unlimited and abnormal over growth of tissues. It is characterized by an abnormal and uncontrolled division of cell exhibiting varying degree of malignancy which produce tumor and invade adjacent normal tissue. It is the second cause of mortality in the world, is continuing to be major health problem worldwide. The currently available nonspecific traditional chemotherapeutic agents only provide palliative care, hence in order to overcome draw backs of chemotherapeutic agents, identification of specific tumor targets and design of novel analogues is very important. Therefore, the development of novel therapeutic agents for the treatment of cancer is of vital importance. Continued development of novel drugs is necessary to improve patient survival and limit treatment-associated toxicities. Whilst the majority of anti-cancer agents are of natural origin, the significance of ‘made to order’ synthetic drugs in the compound against cancer cannot be disputed. Nanoparticles have play important role in treatment of cancer, the advantages of targeting cancer by simply being accumulated and entrapped in tumors (passive targeting). The phenomenon is called enhanced permission and retention effect, caused by leaky angiogenetic vessels, poor lymphathatic drainage and has been used explain why microtubules and nanoparticles are found higher in tumor as compared to normal tissue.
Anticancer agents are used for treatment of cancer, they either kill cancer cells or modify their growth [1]. Drug discovery and developing a new medicine is a long, complex, costly and highly risky process that has few peers in the commercial world. That’s why Computer-Aided Drug Design (CADD) approaches are being widely used in the pharmaceutical industry to accelerate the process. The cost benefit of using computational tools in the lead optimization phase of drug development is substantial. CADD is capable of increasing the hit rate of novel drug compounds because it uses a much more targeted search than traditional high throughput screening (HTS) and combinatorial chemistry. It explains the molecular basis of therapeutic activity and to predict possible derivatives that would improve activity. In a drug discovery campaign, generally CADD is used for three major purposes such as (1) filter large compound libraries into smaller sets of compounds that can be tested experimentally; (2) guide the optimization of lead compounds, to increase its dystrophia myotonica protein kinase (DMPK) properties including absorption, distribution, metabolism, excretion and toxicology (ADMET); (3) design novel compounds, either by “growing” starting molecules one functional group at a time or by piecing together fragments into novel chemo types [2]. It has two types one is structure-based and another is ligand-based. Structure-based CADD relies on the knowledge of the target protein structure to calculate interaction energies for all the compounds to be tested, where as ligand-based CADD exploits the knowledge of known active and inactive molecules through chemical similarity searches or construction of predictive, Quantitative Structure-Activity Relationship (QSAR) models [3]. Numbers of techniques are involved in CADD, from this Molecular Modeling (MM) is one, It can be stated as “MM is anything that requires the use of a computer to paint, describe or evaluate any aspect of the properties of the structure of a molecule” [4]. Methods used in the MM are automatic structure generation, analysis of three-dimensional (3D) databases, construction of protein models by techniques based on sequence homology, diversity analysis, Molecular Docking (MD) of ligands or continuum methods. On the other hand, this discipline includes all methodologies used in computational chemistry, like computation of the energy of a molecular system, energy minimization, Monte Carlo methods or molecular dynamics. We can also say that in another words, it is possible to conclude that computational chemistry is the nucleus of MM [5]. Nowadays another approach proved to be a tool in minimizing the tedious drug discovery process that is Structure Based Drug Design (SBDD). It opens the new world of drug discovery into a target specific path of a drug towards disease.
Computational docking (In Silico molecular docking or just docking) is a computational science aiming at predicting the optimal binding orientation and conformation of interacting molecules in space, and to estimate the stability of their complex. MD predicts whether or not the two molecules interact, the binding affinity and the 3D structure of the complex. It is a key tool in structural molecular biology and CADD. They can screen several compounds for the specific activity within in a limited period of time. It is frequently used to predict the binding orientation of small drugs candidates to their protein targets in order to in turn predict the affinity and activity of a small molecule. The goal of ligand–protein docking is to predict the predominant binding mode(s) of a ligand with a protein of known 3D structure. There are two types of MD such as Flexible docking and Rigid docking, in Flexible docking of ligand to receptor molecules is an emerging approach and is extensively used to reduce cost and time in drug discovery. It is a study of receptor of protein fit together the problem is solving 3D puzzle, it encompasses all theoretical methods and computational techniques used to model or mimic the behavior of molecules. Their software used in the drug development process. It is safe and easy to use tool helps in investing, interpreting, explaining and identification of molecular properties using 3D structures. It may be applied to hit identification, lead optimization, bioremediation, searching for lead structure for protein target, comparing set off inhibitors, estimating influence of modification in lead structures, de Novo Ligand design, understanding the binding principle, optimizing lead structures. Thus MD plays vital role in the new drug discovery process [6]. Over the last few decades, it has been routinely and successfully applied in most pharmaceutical and biotech companies for a large number of applications.
MD classifies biomolecules into three categories such as small molecules (‘ligands’), proteins, and nucleic acids. The most important types of docking systems are protein-ligand, protein-protein and nucleic acid-protein. The interactions between a small molecule and a protein are by far much better understood than those between a protein and a nucleic acid [7].
Software which is used in MD can be categorized based on the following criteria:
1. Molecular representation - a way to represent structures and properties (atomic, surface, grid representation)
2. Scoring method - a method to assess the quality of docked complexes (force field, knowledge-based approach.)
3. Searching algorithm - an efficient search algorithm that decides which poses to generate (exhaustive search, Monte Carlo, genetic algorithms, simulated annealing, tabu search)
Steps involved in MD
Step 1 – Preparation of ligands
- Draw your ligands using a Java app let, upload a single ligand file or multiple ligands.
- Draw chemical structures by MarvinSketch or Chemdraw,
- Upload a ligand in MDL MOL, SYBYL MOL2, PDB, HYPERCHEM HIN or SMILES format.
- Upload multiple ligands in SDF format.
- You can set various parameters during the simulation such as desired pH, structure optimization and partial charge calculations using molecular mechanics or semiempirical quantum chemical methods.
- Set up rotatable bonds and atom types automatically or modify manually.
- Download the attached files in several file formats including mol, pdb, mol2 and pdbqt.
- Organize your ligands into self-defined folders. This way the ligands are saved for later docking calculations.
Step 2 – Preparation of proteins
- Upload protein structures from your files or download them from the Protein Data Bank using Docking Server by providing the entry code or by text search.
- Select the protein chain, heteroatoms, ligands and waters present in the protein pdb file that you want to be included in docking calculation in the process of protein setup.
- Setup the simulation box using one of the following ways:
o select known binding site through a co-crystallized ligand
o select the center of mass of the protein
o select the coordinates of the box center
o select amino acid residues that define the binding site
- Molecular Docking Server calculates necessary map files for each atom type and prepares the input files for docking calculations.
Step 3 – Setup ligand protein docking calculations
- Select a protein and a ligand from your library.
- Modify advanced parameters during the simulation, such as number of runs, number of evaluations etc. Step 4 – Evaluation of results
- Choose an image from the image gallery or render in Molecular Docking Server
- Analyze the secondary interactions between the protein and ligand.
- Create a method section for your reports automatically
Heterocyclic chemistry offers an example for the lack of distinct demarcations; in fact, it pervades the plurality of the other chemical disciplines. Heterocycles are inextricably woven into the life processes. Synthetic chemistry provides cornucopia of heterocyclic systems. More than 90% of new drugs contain heterocycles and the interface between chemistry and biology, at which so much new scientific insight, discovery and application is taking place is crossed by heterocyclic compounds. The majority of heterocyclic compounds and typically common heterocycles fragments present in most pharmaceuticals currently marketed, alongside with their intrinsic versatility and unique physicochemical properties have poised them as true cornerstones of medicinal chemistry. Apart from the already marketed drugs, there are many others being investigated for their promising activity against several malignancies. In particular, anticancer research has been capitalizing on the intrinsic versatility and dynamic core scaffold of these compounds. In the race of new anticancer drug development, heterocyclic derivatives, due to their widespread therapeutic uses, have attracted a great deal of attention amongst the scientific community.
With its origins rooted in organic synthesis and medicinal chemistry, heterocyclic compounds present themselves as a fundamental division of organic chemistry. The type and size of ring structures, together with the substituent groups of the core scaffold, impact strongly on the physicochemical properties [8-10]. Heterocycles can be easily manipulated to achieve the required modification in function and their moieties are part of compounds showing numerous biological activities like antibacterial, anti-viral, anti-fungal, anti-inflammatory, and antitumor Hence, the present study focuses on the SBDD approach for anticancer activity of various substituted Benzothiazole, Thymoquinone, Oxadiazole, Benzthiazine, Carbaldehyde, Triazine, Acetidino-quinazoline, Indolyl Chalcone, Sulfonamides, Coumarin, Thiazolidiones, Darunavir, etc [11,12].
Cancer affects people at all ages with the risk for most types increasing with age. The traditional anticancer drugs are the basis for the, new drug development for cancer in which imidazole is an important moiety. Imidazole is a heterocyclic ring containing basically 3C and 2N atom present in 1st and 3rd positions [13]. The substitution on different positions gives a number of compounds of interest. It has been found that there are a variety of heterocyclic compounds used as anticancer agents. Among the heterocyclic compounds the thiophene, pyridine, thiazole derivatives [14] showed high cytotoxicity towards the cancer cell lines. Presently a number of drugs are used in the treatment of the cancer, but majority of them were produced controlled effect on the cancer cell. By application of these drugs the disease can be controlled. The derivation of Structure Activity Relationships (SARs) is central to MM. SARs are widely used in the systematic design and refinement of pharmaceutical agents and in the identification of structural alerts of toxicity and mutagenicity. Because of their widespread importance both in fundamental and commercial research, many methodologies have developed [15,16]. In this review, we provide for a concise overview of heterocyclic active compounds and families and their main applications in medicine. We shall focus on those suitable for cancer therapy while simultaneously addressing main biological targets, structure-activity relationships as well as MD issues in the use of these compounds.
Main body
Suby T Baby et al have reported that human Heat Shock Protein 90(Hsp90) was involved in many signaling networks associated with cancer progression, so it is an important target for cancer therapeutics. Therefore, they recognized potential lead molecule among the selected heterocyclic compounds against Hsp90 (PDB: 1YET) through MD (GOLD 3.1). A primary objective in MD was the ability to estimate the scoring function and evaluate protein– ligand interactions in order to predict the binding affinity and activity of the ligand molecule. By using a genetic algorithm method to explore the conformational space of the ligands and it give some flexibility of active site residues. Docking calculations were performed using the default GOLD fitness function and their parameters were used to produce the set of optimal conformations of both the ligand and the protein. Greater the GOLD fitness score better the binding affinity. A Pharmacophore study (Discovery studio 2.1) was carried out to construct a hypothetical pharmacophore model aiming to study fitting of the designed compounds to the generated pharmacophores of our target protein Human Hsp90. To build the pharmacophore model for screening, corresponding possible interaction points from active site of Human Hsp90 protein was generated using the ‘Interaction Generation’ protocol implemented in Discovery Studio. It extracted all the available hydrophilic and lipophilic interaction points that can be complemented by the inhibitor. Compounds were ranked based on fit values. A higher fit value represents a better fit. On these basis compounds are analyzed and results of docking and pharmacophore showed that the binding affinity and good fit values of Compound 1 and Compound 2 with our target protein was reliable, showing significant interactions, suggesting their inhibitory activity as shown in Figure (1).
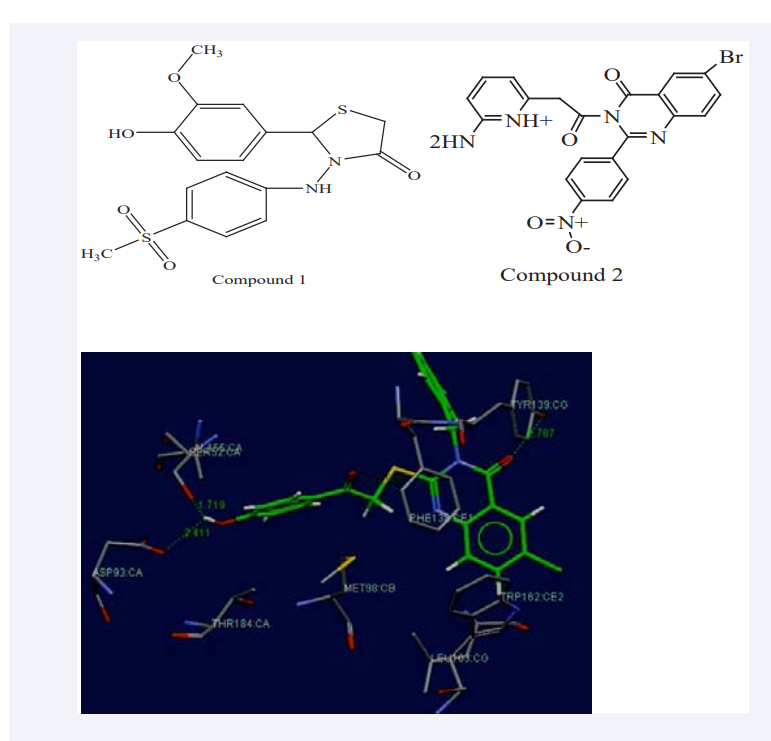
Figure 1: Hydrogen bond interactions of Compound 1 and Compound 2 with Hsp90.
By correlating pharmacophore results with the docking result signifying that both these compounds show antagonistic activity or potential inhibitor of HSP90 [17]. Gajendran nithya et al., have explained the statistics on cancer imposes the urge to extend new methods to control this deadly form of the disease. The Phosphatase and tensin homolog located on chromosome 10q23 (PTEN) is inactivated in a subset and AKT is frequently activated in cancer. The PTEN is the central negative regulator of the phosphatidylinositol 3-kinase (PI3K)/ AKT signaling cascade that influences multiple cellular functions including cell growth, survival, proliferation and migration in a context-dependent manner. Dys-regulation of this signaling pathway contributes to different types of cancers. In that they explore the anti-cancer potential of Thymoquinone (TQ) by analyzing the interaction between TQ with the target protein PTEN. The 3D structure of TQ is designed using in-silico methods and the structure of PTEN is obtained from National Center for Biotechnology Information (NCBI). TQ showed the binding energy of −7.37 Kcal/mol against PTEN with three hydrogen bonds as shown in Figure (2).
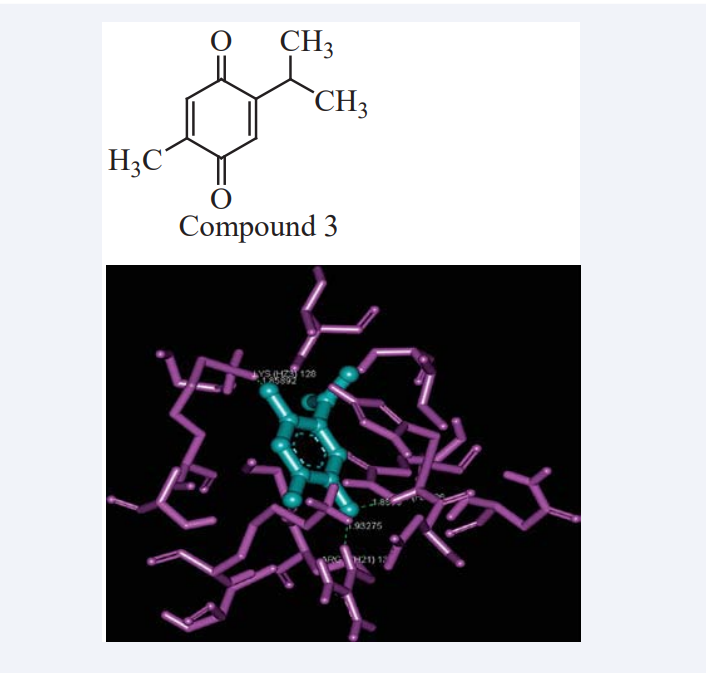
Figure 2: Docked orientation Ligand with PTEN, H-bond formed are denoted by green dotted lines.
They conclude that TQ might inhibit abnormal cell proliferation occurring in cancer by modulating the activity of PTEN, a negative regulator of PI3K/AKT pathway and thereby preventing the cancer initiation and development [18]. Ade Arsianti et al., have reported the synthesis and MD study of analogues of antimycin A3 as inhibitors of anti-apoptotic Bcl-2 of breast cancer. For these purpose twenty designed compounds and the original antimycin A3 were docked (MOE 2009.10 software) based on their interaction with breast tumor receptor binding target Bcl-2 as shown in Figure (3).
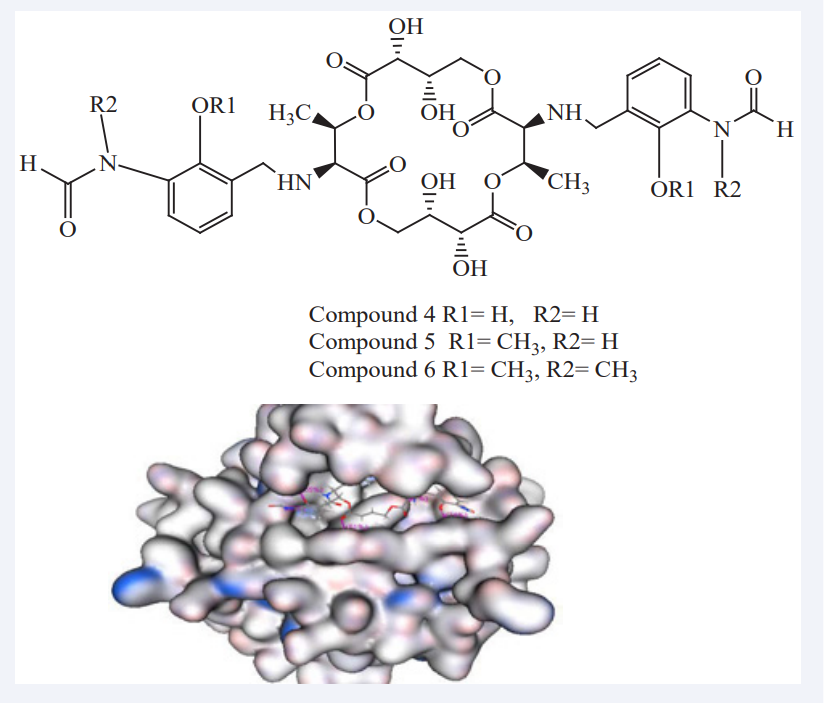
Figure 3: Conformation of compound on catalytic site of Bcl-2 breast cancer.
The five top-ranked compounds with their docking score (London DG) namely, Compounds 4,5 and 6, which have a lower ?G binding energy, better affinity and stronger hydrogen bonding interactions to the active site of Bcl-2 than antimycin A3. The analogue compounds, which have an 18-membered tetraol core exhibited the strongest hydrogen bond interaction, formed high stability conformation, and demonstrated the greatest inhibitory activity on the catalytic site of Bcl-2 breast cancer compared to the original antimycin A3 [19].
Mohan jayapriya et al., have studied a series of benzothiazine compounds for ADME (QuickPro) properties to assess their drug-like properties. These compounds were selected for MD (Glide software) studies as PIM1 kinase inhibitors. Compound 7 showed a Glide score of −7.622 kcal/mol with good hydrophobic and hydrophilic interactions with PIM1 kinase proteins as shown in Figure (4) and appear to be more potent.
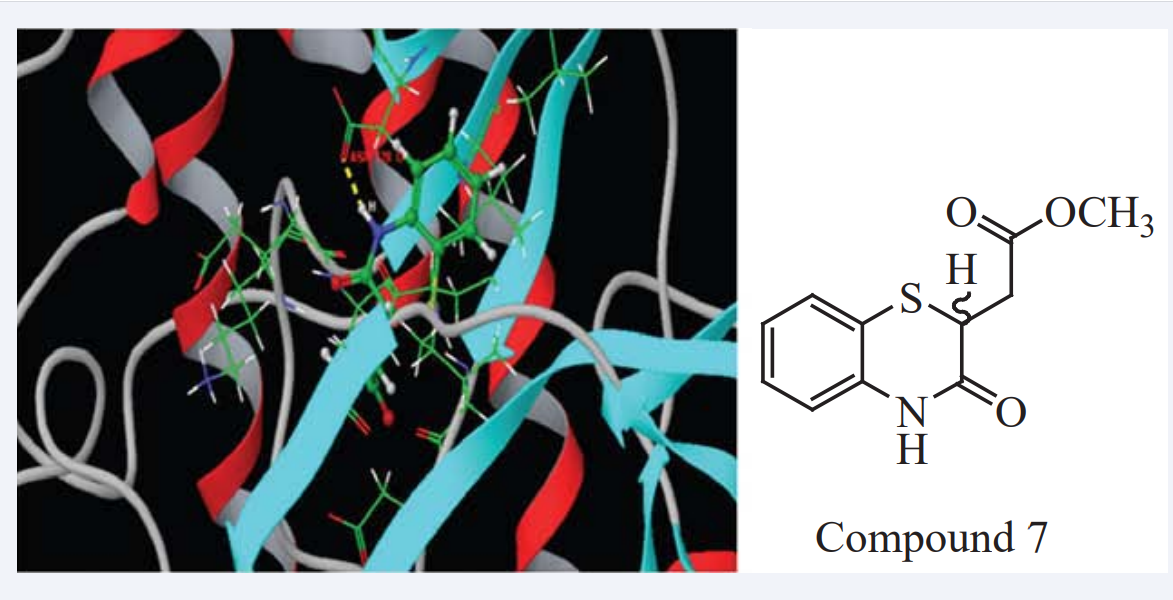
Figure 4: Interaction of target protein PIM1 with Compound 7.
An analysis of the structure - Affinity relationships suggested that the substituent at position three is important in influencing affinity. It was further confirmed by its in vitro anticancer activity of Compound 7 against K562 cell lines by MTT assay by exhibiting an IC50 value of 36.82. Finally they conclude that compounds with an alkyl spacer between the carboxyl group and the core benzothiazine ring are required for binding of the compounds with the PIM1 kinase [20].
Leena. K. Pappachen et al have carried out the docking studies of benzothiazole derivatives containing oxadiazole groups or amino groups with known anti-cancer and anti-inflammatory targets like estrogen receptor, cox1 and cox2 by using Argus lab and Auto dock programmers. Docking studies (Auto dock versin 4.0) done on all ten ligands and docking scores were compared with the scores of standard drugs Tamoxifen and Indomethacin. Validation of ligands was carried out by using Lipinski rule of five. All these ligands show higher docking scores and shows better drug likeness properties as compared to the reference drugs. All the compounds show lowest docked energy and hydrogen bonding stabilizes the interactions as mention in Figure (5).
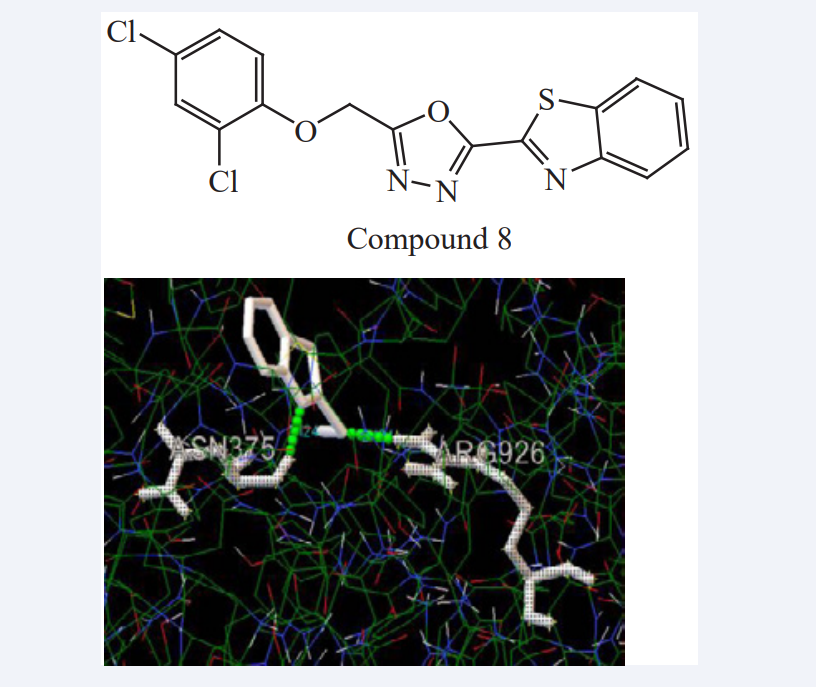
Figure 5: Compound 8 Cox1 (1cqe) hydrogen bonding interactions (ASN 375, ARG 926) (-5.93 kcal/mol).
The analogues of benzothiazole at second position showed good receptor binding with the selected targets. Substitution on 5th position of oxadiazole ring with aromatic group also increases the activity. Introduction of an ethylene bridge between oxadiazole and aromatic substitution resulted in an analogue with best binding potency. The final assessment of drug-likeness and its related parameters helps to confirm the oral activity of compounds [21].
Deepak Kumar Basedia et al have reported a series of compounds 1,3,4-oxadiazolo-[3,2-a]-1,3,5-triazine and 1,3,4-thiadiazolo-[3,2-a]-1,3,5-triazine derivatives which were successfully synthesized and their structures were confirmed by spectral analysis (IR, NMR & Mass). MD (V-Life MDS) of title compounds were done on breast cancer protein BRCA1 (PDB: 2IOK) and their protein receptor interaction study has carried out to identify potential anticancer compound based on dock score. The Compound 9 show promising dock value -70.24. Compound bind with protein BRCA1 (PDB: 2IOK) by forming hydrogen bond interaction, hydrophobic interaction with amino acid residues as shown in (Figure 6). To strengthen the current investigation, further evidences both in vitro and in vivo are needed so as to use this approach effectively for cancer treatment [22].
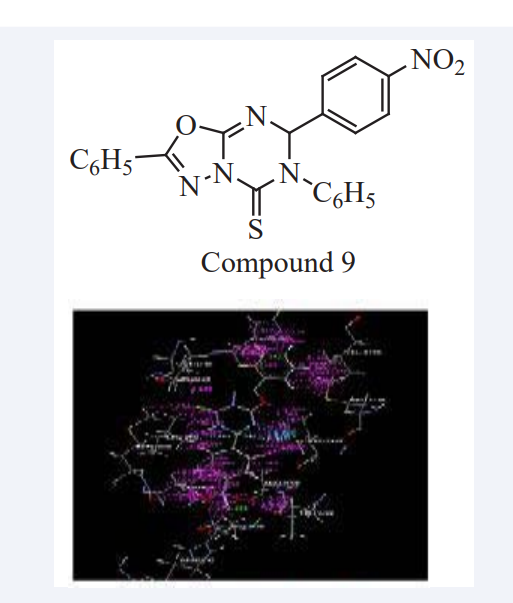
Figure 6: Graphical interpretation of protein BRCA1 with Compound 9.
Ayyappan Aravind et al have focused on the SBDD and development of novel Acetidino-quinazoline derivatives and their antiproliferative evaluation. The compound that inhibits the epidermal growth factor receptor (EGFR), a tyrosine kinase receptors are directly related to blockade of regulatory processes of cellular proliferation hence it is suitable targets for cancer treatment. They were carried out In-silico design (ACD labs ChemSketch 12.0) and analyze ‘Lipinski Rule of Five’ and drug likeness properties (Molinspiration) of novel analogues. Biological activity was predicted by PASS software. After in-silico design all the proposed derivatives were subjected to flexible docking on to the binding pocket of EGFR (Pdb ID: 1M17) using GLIDE program of Schrodinger as shown in Figure (7).
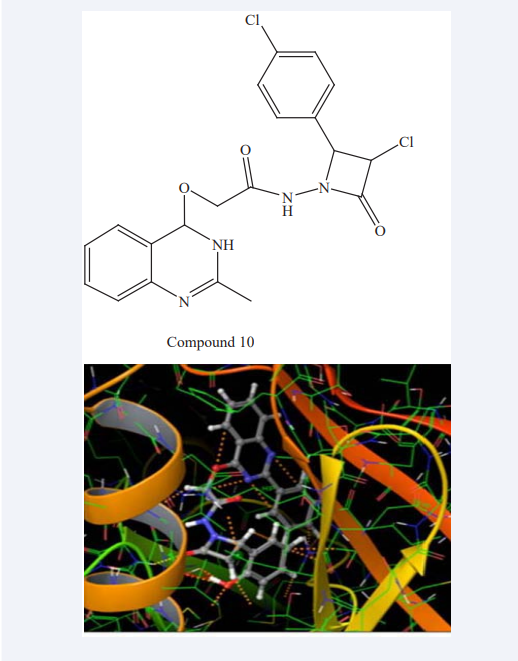
Figure 7: Docking images of Acetidino-quinazoline derivatives Compound 10 on binding pocket of EGFR.
Then the selected five analogues were synthesized by wet lab synthesis. These derivatives were spectrally characterized by FT-IR, 1HNMR, MS. The Biological evaluation was done by MTT assay using human cervical cancer cell lines (HeLa). The Compound 10 have shown significant activity against HeLa cells and compared with that of standard drug Paclitaxel then they conclude that the compound can be lead candidate for drug discovery. This will lead to the development of promising lead compounds for target specific anticancer therapy and encourage further optimization to develop potent anti proliferative agent [23].
M. Yaseen Mowlana et al have reported according to Claisen-Schmidt condensation a series of novel substituted Indolyl Chalcone derivatives were synthesized and characterized by TLC, elemental analysis, IR and 1H and 13C NMR spectroscopy and evaluated docking study [24] that were subsequently screened for activity against bacterial and fungal strain. In that the docking study involves protein along with the BRCA1 receptor, in that one hydrogen bond proves to be a most stable complex inhibiting the activity of integrase as shown in Figure (8). For evaluating accuracy of docking, the binding energy and numbers in cluster was used. The interaction shows that it binds to the protein and specifically interferes with its strand transfer activity [25].
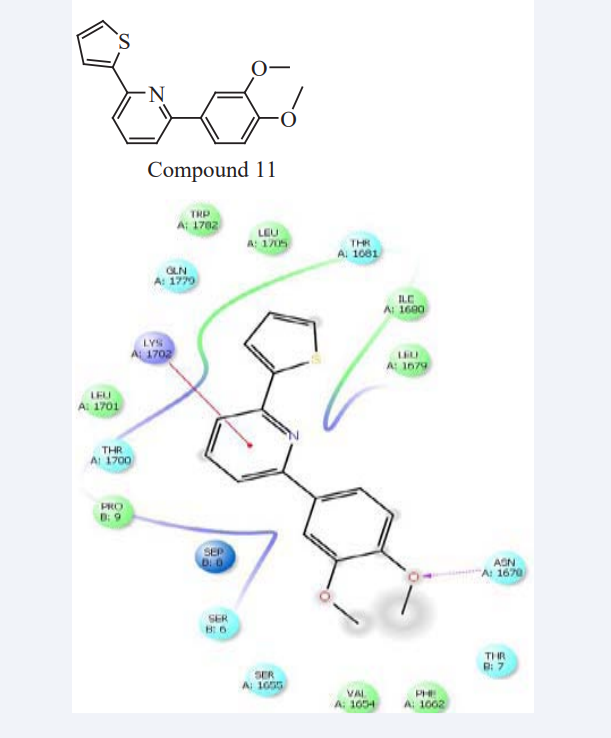
Figure 8: Binding mode analysis of Compound 11 Derivatives with Target protein BRCA1.
Sohrab Ghanei et al., have stated that carbaldehydes received considerable attention owing to their synthetic and effective biological importance which exhibits a wide variety of biological activity. A group of N1, N4-bis ((2-chloroquinolin-3-yl) methylene) benzene-1, 4-diamine derivatives were synthesized, and theoretically evaluated for their inhibitory as Potential Human AKT1 Inhibitors via docking process (GOLD 5.2.2). Those derivatives were synthesized from 2-chloroquinoline-3-carbaldehydes. As the inhibitor of AKT1 (RAC-alpha serine/ threonine-protein kinase is an enzyme that in humans is encoded by the AKT1), the aforementioned compounds may have implication in preventing complications of cancers. The docking was carried out and Compound 12 showed the best inhibitory potency by GOLD score value of 113.76. The compound formed strong hydrogen bonds with Asn 49, Lys 220, Ser 157, Arg 225 and Trp 76 via quinoline moiety and nitrogen of quinolone ring. Pi-pi interaction between Lys 220, Trp 76, Tyr 224, Arg 225, Ile 80, and Asn 49 quinoline moiety was one of the common factor in enzyme-inhibitor junction as mention in (Figure 9). It was found that both hydrogen bonding and hydrophobic interactions are important in function of biological molecules, especially for inhibition in a complex [26].
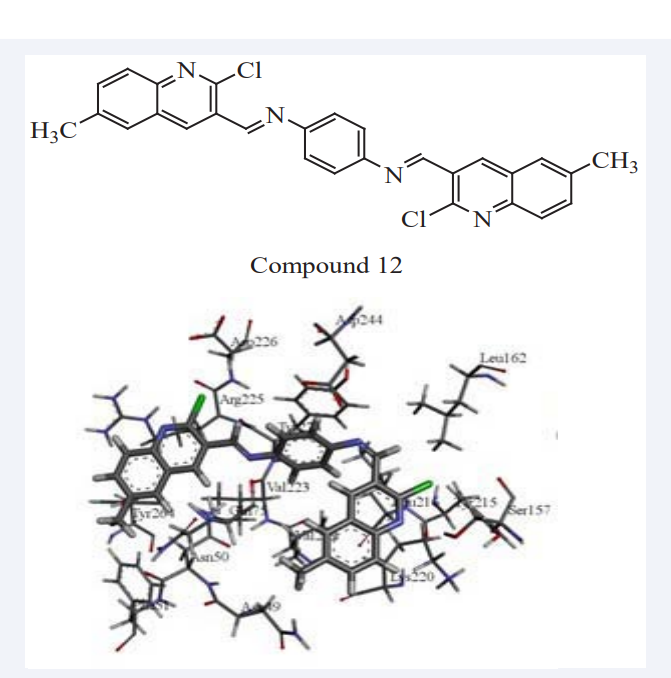
Figure 9: The Pi-pi interaction and hydrogen bonding of Compound 12 with the active site pocket of AKT1 in stick
Mahmoud Sayed Bashandy has described the synthesis of some novel sulfonamides having the biologically active, thiazole, imidazo[1,2-a]pyridine, imidazo[2,1-c] [1,2,4] triazole, imidazo[2,1-b]thiazole, nicotinonitrile, 1,3,4-thiadiazine, quinoxaline and 1,4-thiazine moieties, in this 1-(4-(pyrrolidin-1-ylsulfonyl)phenyl)ethanone used as a starting material [27]. Molecular Operating Environment (MOE) as the computational software, it performed virtual screening using MD studies of the synthesized compounds. All the minimizations were performed with MOE until a root mean square deviation (RMSD) gradient of 0.05 K Cal/mol?Å with MMFF94X force field [28]. The results indicated that some synthesized compounds are suitable inhibitors against dihydrofolate reductase (DHFR) enzyme (PDB ID: 4DFR) with further modification. MD studies further help in understanding the mode of action of the compounds through their various interactions with the active sites of DHFR as shown in (Figure 10), identical charges were automatically calculated. Compounds were tested in-vitro antihuman liver hepatocellular carcinoma cell line (HepG2) exhibited better activity than methotrexate as a reference drug [29].
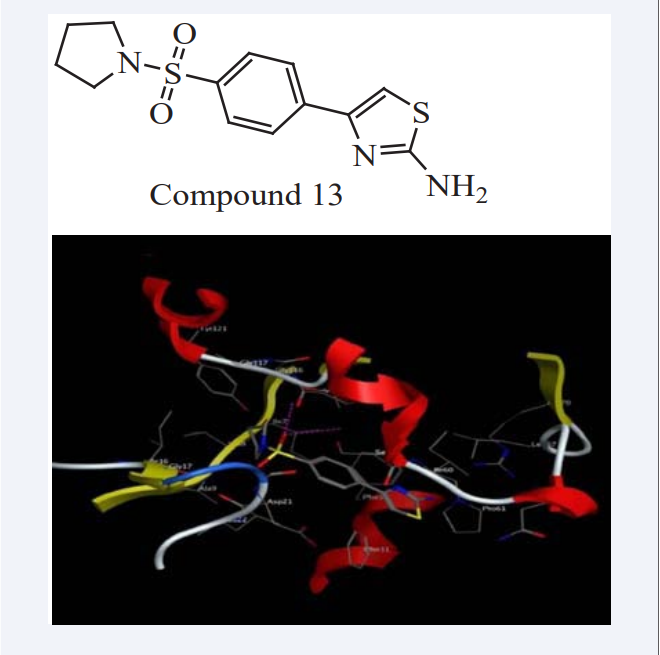
Figure 10: Docking of Compound 13 with DHFR inhibitors.
Sobhi M. Gomha et al have reported reactions of ethylidene thio carbohydrazide with hydrazonoyl halides gave 1,3-thiazole or 1,3,4-thiadiazole derivatives. The cytotoxic activity of the selected products against the Hepatic carcinoma cell line (Hepg-2) was determined by MTT assay. The cytotoxic effect was due to its inhibitory effect to the inner mitochondrial membrane lactate dehydrogenase enzyme (LDH-5), which, in turn, decreases cellular activity, including the rate of cell division. It has been reported that LDH-5 has an important role in tumor maintenance in many human cancers like the hepatic cancer [31] which confirms that LDH-5 inhibition is a good target for developing anticancer agents. The binding mode of the active compounds was interpreted by a MD (Leadit 2.1.5). They were performed this study to interpret the LDH inhibitory activity for the synthesized compounds that were confirmed biologically. A direct correlation between the activity and the binding affinity was observed also the entropy ligand conformation score for the highly active group was a neglected value (0.00), which is favored for best fitting. The MD study confirmed high binding affinities of –24.85, –kcal/mol for Compounds 14. Thiazol-2-(3H) moiety was important to form a hydrogen bond with Thr 99, Asn 113 and Ala 96 as mentioned in (Figure 11). The N=N and C=N groups were involved in the interactions. An overall good fitting and placement was observed for the docked compounds inside the active site where the nicotinamide-adenine dinucleotide was bound [30].
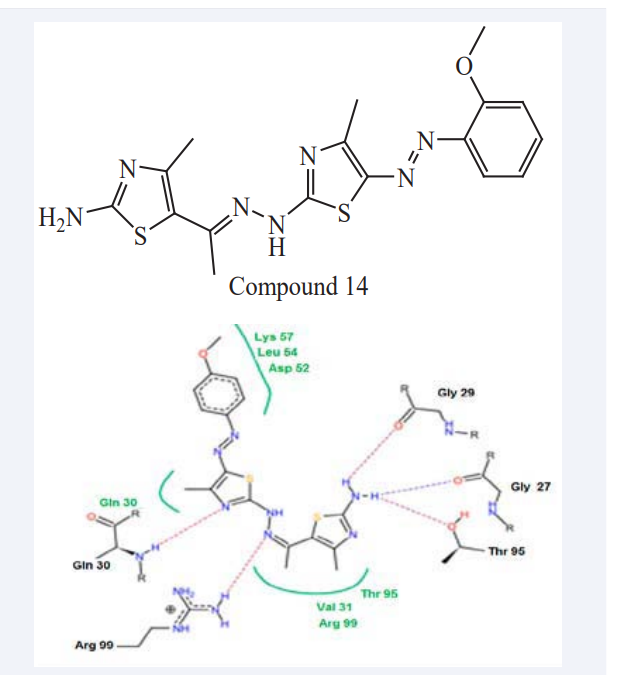
Figure 11: Possible binding modes of Compound 14.
Elgemeie GH et al have synthesized a novel series of 2-thioxoimidazolidin-4-one and Benzothiazole thioglycosides via one-pot reaction. The cytotoxic activity of compounds were evaluated against MCF-7 cell lines (Breast carcinoma cell lines) showing high to moderate anti-tumor activities moreover MM of these compounds revealed that they have high binding affinity through hydrophobic-hydrophobic interaction and moderate selectivity through the hydrogen bond interaction with the atypical nucleotide binding pocket in the amino terminus of HSP90 as shown in (Figure 12). After MD result it confirmed that Compound 15 has Consensus Score 431 and Novobiocine have a Consensus Score 538, hence it could be potential drug candidates for cancer treatment [31].
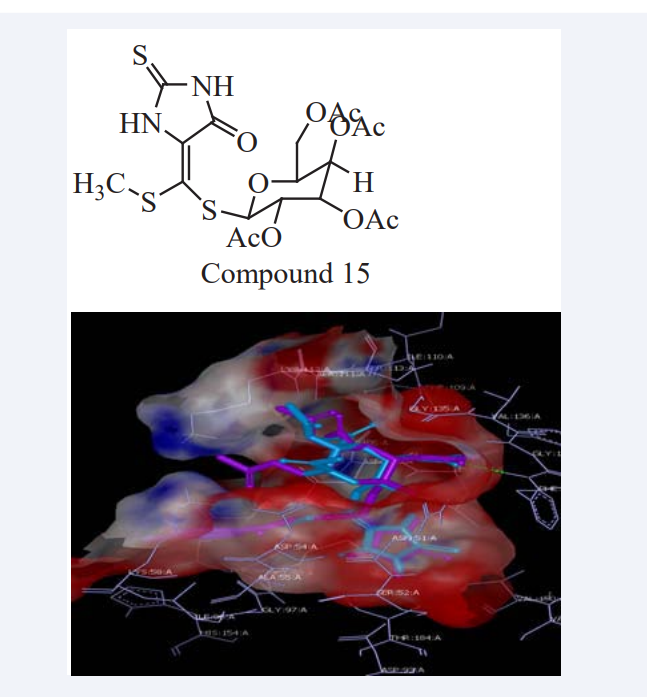
Figure 12: Binding-site analysis and its graphical interpretation.
Francis M Saleshier et al., have done CADD to bring out the molecular study of the enzymes Topoisomerase II inhibitors and EGFR TK inhibitor, they are used as anticancer drug for decades and works by stabilizing the enzyme induced DNA breaks. In that docking (Auto dock 4.2 version, Acccelrys Discovery Studio, and Cygwin) and Virtual screening (iGEMDOCK and molinspiration server) results confirmed the possibility of benzothiazole moiety possessing the anticancer activity. The PDB accession code for EGFR is 21TY and for topoisomerase II is 2RGR. In the present work the tumorigenic cell line activity of the substituted benzothiazole is compared with the two standard drugs Gefitinib and Doxorubicin by an In-vitro assay against Dalton’s Lymphoma Ascites cell using trypan blue dye and percentage inhibition was calculated. The predicted activity and In-vitro activity of benzothiazole for topoisomerase II inhibition were easily correlated. Compound with lowest binding energy was having the highest binding affinity towards the target 2ITY and 2RGR as shown in Figures (13,14) respectively. Binding energy of Std -1 docked with 2ITY is -8.53 and the binding energy of Std 2 docked with 2RGR is -8.86. It has proven as a potent anti-cancer agent, via topoisomerase II inhibition and EGFR- TK inhibitor[32].
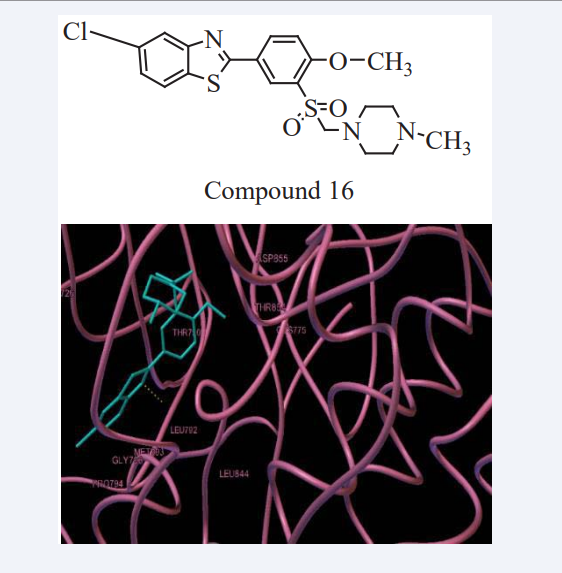
Figure 13: Ligand docked to 21TY.
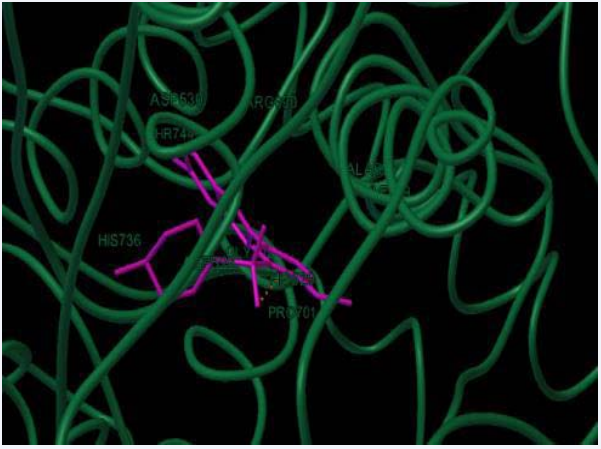
Figure 14: Ligand docked to 2RGR.
Jagadish Tota et al., have presented work, in that a new Schiff base ligand and its metal complexes were synthesized, such compounds were analyzed through various spectral techniques. The analytical data has shown the metal to ligand ratio is 1:1. IR spectral studies revealed that binding sites of ligand with metal ions through the azomethine nitrogen and imidazole nitrogen atoms. The core intension of the present work is to check the biological activities of the compounds. All the compounds were screened against bacterial strains and HeLa, MCF-7 cancer cell lines. Metal complexes have shown good antibacterial activity than ligand, but in case of anticancer activity both Schiff base ligand and its metal complexes have shown greater activity. The binding mode of the ligand with protein active sites were predicted using MD technique. Interaction of docked pose of ligand in the protein active site showing hydrogen bond interactions with PHE1 60 as shown in (Figure 15). The dock score values of ligand showed a correlation with the inhibitory activity against Staphylococcus aureus pencillin binding protein 4, E. Coli Penicillin Binding Protein 4 (dacB) and Homo sapiens cyclin dependent kinase [33].
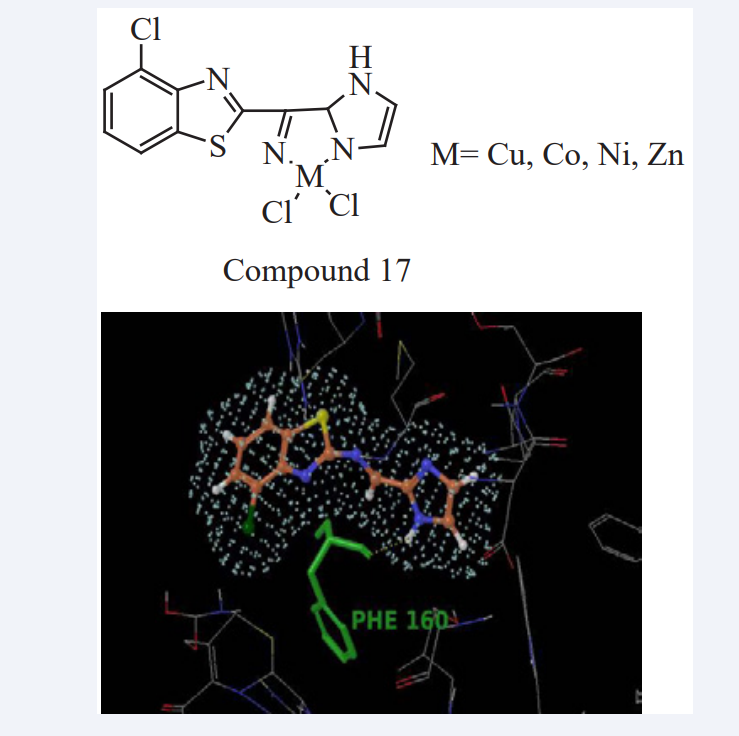
Figure 15: Docked pose of ligand in the protein active site showing hydrogen bond interactions with PHE160 in E.Coli.
Shiny George et al have computationally designed a series of coumarin derivatives and optimized (AutoDock 4.0.1) to investigate the interactions between the target compounds coumarin derivatives and the amino acid residues of the N-acetyltransferase 2 (NAT2) receptor. Among all the designed compounds Compound 18 shows more binding energy values -9.08, It binds with 2PFR along with interacting amino acid as shown in (Figure 16). It indicates that coumarin derivatives are excellent promoters of NAT2. It is found to be most active against Arylamine N-acetyltransferase 2. Coupling of these compounds with other known anti neoplastic natural products for the finding of more promising anti-colorectal cancer compounds are being analyzed [34].
Figure 16: Binding mode of Compound 18 in the active site of 2PFR along with interacting amino acid.
Mohamed Jawed Ahsan et al have synthesized a series of new quinoline analogues in two steps. The Compound 19 showed maximum docking score and maximum anticancer activity with GI50 of 35.1μM against HeLa (cervix cancer cell line) and 60.4 μM against MDA-MB-435 (breast cancer cell line) respectively. A MD study implying EGFR-TK was carried out to observe the binding mode of new quinoline analogues on the active site of EGFR-TK. The amino acid residues Met793 showed backbone H-bonding with the hydroxyl group, while Asp855 showed side chain H-bonding with aryl NH group as shown in Figure 17. This compound could be considered as lead for further optimization and drug discovery. The quinoline derivatives discovered in this study may provide valuable information in the field of drug design and cancer therapy [35].
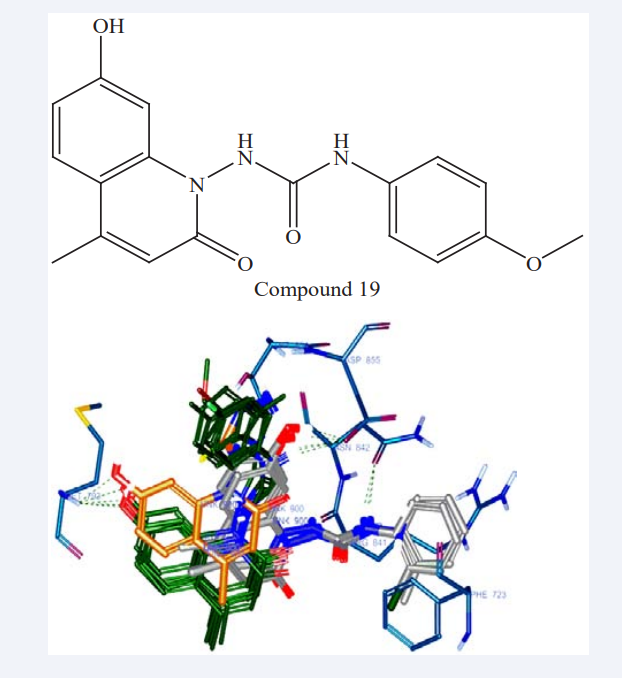
Figure 17: The binding mode of Compound 18 with EGFR tyrosine kinase active site.
Metta Madhuri et al., have studied the ligand-protein MD (Molegro Virtual Docker v 5.0.) simulation was used to preliminarily investigation and to confirm the potential molecular target for the thiazolidinediones compounds with observed cytotoxicity. The analysis of the best docked ligands against selected anticancer drug target revealed the binding mode of compounds involved in this study and confirm the role as EGFR inhibitors. Binding energies of the drug–enzyme (receptor) interactions are important to describe how fit the drug binds to the target macromolecule. The residues participated in the hydrogen bond formation within the active binding site region revealed the importance of these residues towards the observed binding energy with respect to the hit identified against EGFR target protein as mention in Figure 18. The obtained hypothesis could be the remarkable starting point to develop some new leads as potential EGFR inhibitors with enhance the affinity as well as intrinsic activity. The results of this work indicate efficient computational tools are capable of identify potential ligand such as Compound 20 which was earlier reported in our work as potential cytotoxic agent [36].
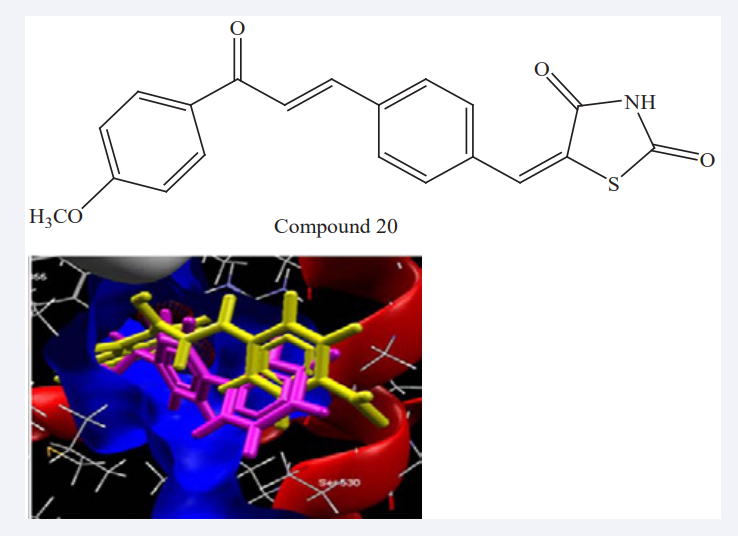
Figure 18: Superimposed binding orientation of docked conformer and most stable ligand within the active binding site region of 1M17.
Jhinuk Chatterjee et al., have studied Cardamom (Elettaria cardamom), have a cancer chemo preventive potential in in-vitro and in-vivo systems. In that they has been made, a fruitful attempt to characterize the proapoptopic, anti-inflammatory, anti-proliferative, anti-invasive and anti-angiogenic targets of bioactive principles in cardamom using comparative Inverse screening approach using Reverse Screen 3D and Pharm Mapper. Interaction studies of binding pose of eucalyptol with Caspase 3(RCSB) were conducted to obtain an insight into the interacting amino acids and their inter-molecular bondings, it is nucleophilic attacks by polar residues inside the Caspase 3 catalytic site. A prioritized list of target proteins associated with multiple forms of cancer and ranked by their Fit Score (Pharm Mapper) and descending 3D score (Reverse Screen 3D) were obtained from the two independent inverse screening platforms. MD(AutoDock Vina in PyRx 0.8) studies exploring the bioactive principle targeted action revealed that H-bonds and electrostatic interactions forms the chief contributing factor in inter-molecular interactions associated with anti-tumor activity [37].
Matcha Bhaskar et al., have reported Darunavir is a synthetic non peptidic protease inhibitor which has been tested for anticancer properties. These analogues act by obstructing the activity of other signaling pathways which are implicated in many tumors. QikProp result state that all the analogues show specified range of all the pharmacokinetic properties required to become the successful drug. MD study was performed to test its anticancer activity against the biomarkers of the five main types of cancers i.e. bone, brain, and breast, colon and skin cancer. They have interacted to all the five biomarkers with sufficient selectivity, specificity and reflected the optimum binding energy to form the stable docked complex. In that the binding model of best scoring analogue with each protein was assessed from their G-scores as shown in Figure (19), and disclosed by docking analysis using the XP visualizer tool. An analysis of the receptor-ligand interaction studies revealed that these nine Darunavir analogues are active against all cancer biomarkers. Most of the analogues have better binding energy to onco proteins with compared to Darunavir [38].
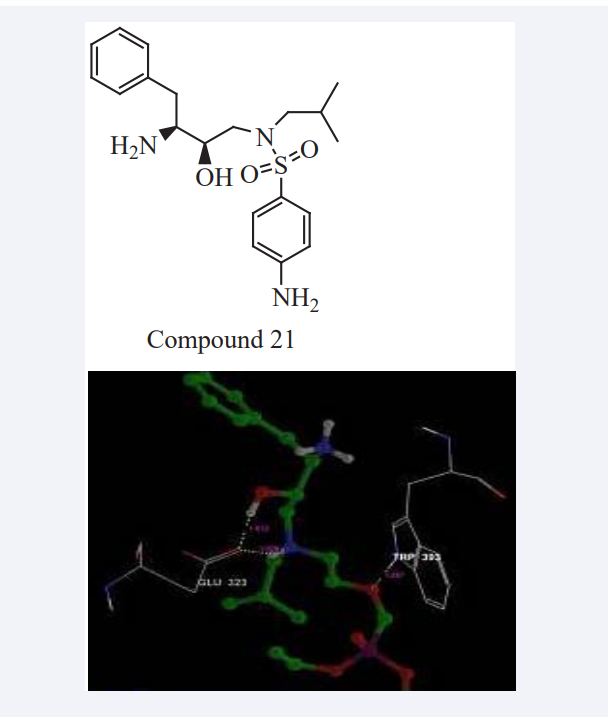
Figure 19: Docking of protein with best ligands.
Akrimah et al have studied Cyclin-dependent kinases (CDKs) are regulatory protein kinases which involved in cell cycle control hence many CDK inhibitors have studied for anticancer potential. Here they conducted a docking (Arguslab CDK 4.0.1) study of 4-(pyrazol-4-yl)-pyrimidine derivatives as CDK1/2 and CDK4/6 inhibitors. In that they compared the docking result of the parent compound, the most selective, and the least selective compound. Interpretation of result in the form of three docking parameters such as- Gibbs free energy(ΔG), atoms and residue of receptor involved in hydrogen bonding, and the bonding length. All compounds had value of ΔG <0, indicated that the interaction between the ligand and receptor was spontaneous. Docking programs are preferred to predict experimental poses with average deviations from 1.5 to 2 Å rmsd [39]. However, this has been an issue for the available docking programs. In some reports, rmsd in range 1.5-3.5 Å is still acceptable [40]. Free energy change (ΔG) is a parameter that indicates the affinity and stability of the interaction between the ligand to the receptor. ΔG value 0 indicates no interaction can take place spontaneously. There is no particular order of these parameters that can be associated with their selectivity order. However, there is a distinction of the residue of the receptor CDK2 that involved in hydrogen bonding as shown in (Figure 20).
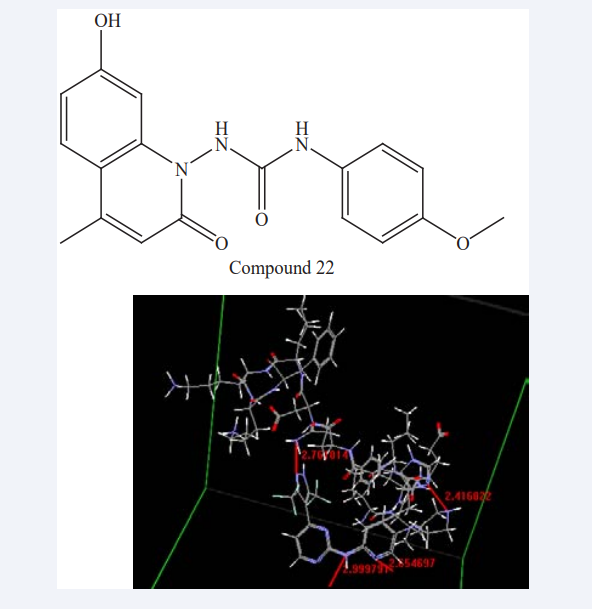
Figure 20: Docking result of ATP binding site at CDK2 into the most selective Compound 22.
The order of the bonding length at 84 HIS residue reflects the selectivity of the compound, the more selective compound has the shorter bonding length. The QSAR equations for CDK2 and CDK4/6 includes AM1_E (total energy), and ASA_H (hydrophobic surface area) descriptors. The distinction between the two QSAR equations was that CDK2 equation contained descriptor molar refractivity (mr), while CDK4 contained polarizability. Molar refractivity is a measure of the steric bulk of a molecule and the value is proportional to the molecular weight of the compound, while polarizability indicates atomic or molecular charge distribution of a molecule [41,42].
Da-An Qin et al have reported two derivatives of 5-fluorouracil (5-FU) were synthesized, and their electrochemical behavior with calf thymus-DNA (CT-DNA) were studied using cyclic voltammetry at a DNA-modified gold electrode. The variations in the cyclic voltametric behaviour of different concentrations of drugs with CT-DNA have investigated. MD was used to predict the modes of interactions of the drugs with DNA and results indicated that it can be considered as groove binding which binds to the minor groove of DNA double helix as shown in (Figure 21).
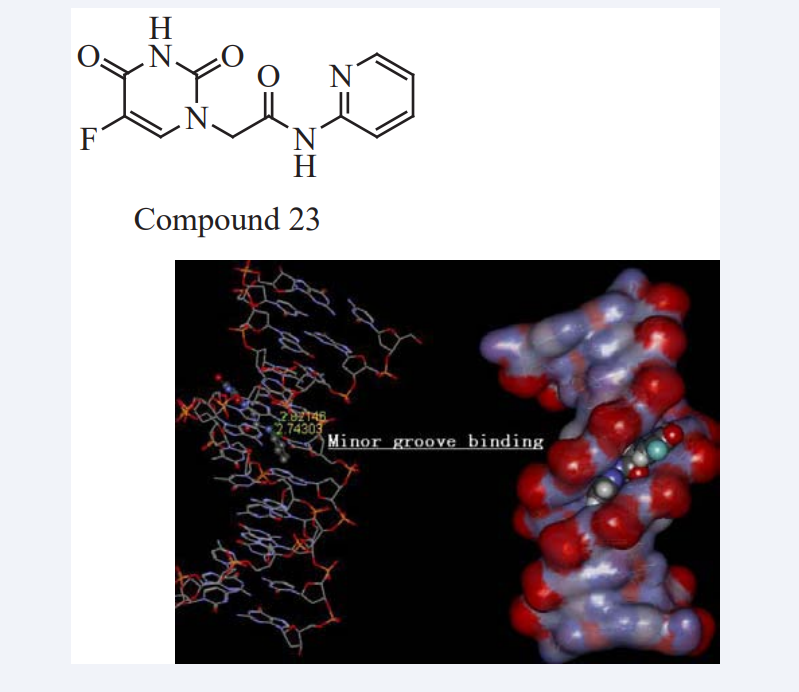
Figure 21: Binding of Compound 23 to DNA.
To measure the binding ability of the two drugs with DNA, binding constants (K) were calculated from voltametric data, i.e., and the binding free energies of the two drugs with DNA were calculated using the AMBER software package. From the binding constants and the binding free energies, it can be concluded that the binding strength of the ortho compound is stronger than that of the meta compound. Therefore, they can make a bold guess that the ortho compound is a potential anticancer drug, which should be used in optimizing the design of this class of 5-FU antitumor drugs [43].
Karpakavalli m. et al have reported curcumin, a phyto constituent of Curcuma longa has pursued the attention of research chemists, as it is found to possess a number of pharmacological activities. In that 4-(4’-hydroxy, 3’-methoxy) phenyl, but-2-one-3-ene, a curcumin analog precursor is aimed to be synthesized and tested for anticancer activity. Aldol condensation of mono carbocyclic aldehyde and the enol form of a 2, 4-diketone in the presence of an organic amine catalyst, the principle is used for the synthesis of said molecule. The invention also relates to the use of the synthesized product for in vivo acute toxicity and in vitro anticancer activity. EGFR were selected from RCSB protein data bank, based on the presence of ligand, X-ray diffraction and 2.0-2.5 A ° resolution. A comparative protein- ligand dock analysis was performed using 2GS6 extracted from PDB to evaluate the algorithm and scoring function efficiency between Auto Dock 4.0.1 and experimental activities as shown in Figure 22. All these computationally designed molecules, as well as the bound ligand of the protein 2GS6, were docked by using the software Auto Dock and the score values are predicted [44].
Figure 22: Protein and ligand (the newly designed molecule) interacted matrix.
In this review article, we have successfully arranged Molecular Docking scores of various heterocyclic compounds that are used in the treatment of cancer. MD gives clues for designing new molecule along with their increased activity and their mechanism of action. We have also depicted the general structure of such potent heterocycles which shall be helpful to the researchers to carry out further research on various structural modifications in these compounds in order to improve the concerned biological activity. Throughout the review, we have focused on the methodologies and software in constructing docking model, namely the Auto dock, V-life MDS, Schrodinger, Pymol, Sybil, Leadit and Gold.
ACKNOWLEDGEMENT
The authors are thankful to Mrs. Fatma Zakaria, Chairman of Maulana Azad Education Trust and Dr. Zahid Zaheer, Principal Y.B. Chavan College of Pharmacy, Aurangabad for their moral support and cooperation.
REFERENCES
6. Mukesh B and Rakesh K. Molecular docking?: A review. Int. J. Res. Ayurveda Pharm. 2011; 2: 1746-1751.
7. Sanschagrin P. An Introduction to Molecular Docking. Structural Bioinformatics (C3210). 2010.
8. Gomtsyan K. Heterocycles in drugs and drug discovery. Chem. Heterocycl. Compd. 2012; 48: 7-10.
11. Arora P. Int J Pharm Res Sci. 2012; 3: 2947.
16. Mohareb RM. Heterocyclic Compounds as Anticancer Agents. Journal of chemical bonding 206: 2014; 1-2.
40. Kubinyi H. Hansch Analysis and Related Approches. VCH. 1993; 95-69.









































































































































