In-silico Identification of Drug-Like Compounds for Targeting HIV-IN and Human Host LEDGF or p75 Interaction
- 1. Department of Biochemistry, Abdul Wali Khan University Mardan, Pakistan
ABSTRACT
The HIV-1causes life-threatening infection by establishing essential biochemical interactions via its IN protein with human host LEDGF/p75 protein. The crystal structure availability of LEDEF–HIV-IN complex provide essential roadmap for designing small drug-like molecule to perturb pathogen-host proteins interaction. We adopted the in-silico drug discovery approaches and scanned the TIMBAL database which hold drug-like molecule promising for protein-protein interactions perturbation. We targeted the interactive hotspot region of the complex to build a pharmacophore model. Pharmacophore based virtual screening revealed single lead compound from TIMBAL repository with reasonable pharmacophore fit score. The drug-like ability of this lead compound along with additional source anti-HIV compounds are examined through molecular docking approaches. We assumed that the putative lead compound addressed in this study will provide a template for identification of additional drug-like molecules for the same purpose in future. The experimental evaluation of lead compound may important for future anti-viral drug discovery.
KEYWORDS
• HIV
• In-silico drug discovery
• Virtual screening
CITATION
Aslam M, Shehroz M, Gul S, Shah M, Khan A (2017) In-silico Identification of Drug-Like Compounds for Targeting HIV-IN and Human Host LEDGF/p75 Interaction. J Bioinform, Genomics, Proteomics 2(1): 1016.
ABBREVIATIONS
LEDGF: Lense Epithelium Derived Growth Factor; HIV-1: Human Immunodeficiency Virus type1; IN: Integrase; CCD: Catalytic Core Domain; IBD: Integrase Binding Domain; PPI: Protein-Protein Interaction
INTRODUCTION
The human immunodeficiency virus (HIV) is retroviruses and causative agent of AIDS. The HIV infection mechanism include the reverse replication of its RNA genome into a double-stranded linear DNA and integration into human host chromosome. The HIV integrase (IN) is a 32-kDa protein responsible for the catalysis of the insertion of viral DNA into the host chromosome [1]. The p75 is form of human cellular protein of lens epithelium-derived growth factor (LEDGF) is an important binding partner of human immunodeficiency virus type-1 (HIV-1) integrase (IN). The LEDGF binds HIV-1 IN via a small, approximate 80-residue IN-binding domain (IBD) within its C-terminal region [2,3]. The IBD is both necessary and sufficient for the interaction with HIV-1 IN [2]. The HIV-IN and LEDGF/p75 protein-protein interaction (PPI) perturbation is been exploited as an important anti-viral target. The complex crystal structure availability (PDB ID: 2B4J) inferred a drug-like cleft in the interface of HIV-IN and LEDGF/ p75 [4]. This provides important platform for structure based drug designing for disruption the biological interaction between HIV and LEDGF/p75 proteins.
The in-silico drug discovery strategies of structure-based pharmacophore modeling, molecular docking and drug-like compounds database virtual screening provide important rationale of drug discovery projects. More specifically, these approaches create computational molecular models and perform high-throughput screening of large databases containing million of compounds to identify lead molecule for desired biological activity and hence ultimately provide in-sight in medicine and therapeutics discoveries [5]. The molecular docking approach model the interaction between small molecule and target protein at the atomic level. This allows characterizing the behavior of small molecules in the binding site of target proteins to elucidate the fundamental biochemical processes [6]. These advances allow the computational strategies to permeate all aspects of drug discovery today. We here adopt these in-silico drug discovery approaches for identification of small molecule potent for targeting HIV-IN and human LEDGF/p75interaction inhibition. Though several reports are available regarding small molecules based inhibition of protein-protein interactions between HIV-IN and LEDGF/p75 [7,8] but none of the so far published studies scanned the TIMBAL repository that hold drug-like compounds specifically potential for protein-protien interaction inhibition.
MATERIALS AND METHODS
HIV-IN–human LEDGF/p75 complex structure retrieval
The HIV-1 IN and human LEDGF complex X-rays diffraction structure of 2.02 Å resolution was retrieved from protein data bank with under PDB-ID: 2B4J [4].
Complex structure protonation and energy minimization
The Molecular Operating Environment (MOE) tool was employed for the complex structure energy minimization and protonation. This aid in the proper orientation of the hydrogen atoms suitable for downstream analyses. Energy minimization of the target proteins were performed at root mean square gradient below 0.05 using MMFF94X force field method implemented in MOE tool. The initial and final energy of the target proteins were calculated (in kcal/mol). The energy minimized structures are used as template for molecular docking analysis. The MOE package based on MM-GB/VI (Molecular Mechanics, Generalized Born Volume Integral) model for binding affinities prediction [9].
Pharmacophore model generation
The structure based pharmacophore model was generated based on the selective hotspot interactive residues of the individual protein within complex. Pharmacophore model based screening is computationally efficient and well suited for virtual screening of large compound libraries. The LigandScout3.12 is recommended tool for interaction detection and pharmacophore model built-up based on complex structure features [10]. An advance alignment algorithm of LignadScout overly the pharmacophore and test compound to infer the common binding modes and shared chemical features. The generated pharmacophore model was satisfied for all essential chemical features.
Virtual screening of TIMBAL database
Virtual screening is in silico method for selecting promising compounds from chemical databases [11]. It can be regarded as the computational counterpart of experimental evaluation of compounds libraries as high-throughput screening (HTS) [12]. TIMBAL database containing 14,890 data points for 6,896 distinct small molecules of molecular weight <1200 Daltons potential to modulate protein–protein interactions [13]. Pharmacophore-based virtual screening of TIMBAL compound repository was done to obtain hit compound against shared features of pharmacophore. The compounds library preparation and virtual screening was done using the internal features of Ligandscout3.12 .The hit compound further examined for drug-like features as according to basic Lipinski rule of five with hydrogen bond donor (HBD) less than 5, hydrogen bond acceptor (HBA) less than 10 and molecular weight no more than 500 Da and logP ranges between 0-5 [14].
Molecular docking
Molecular docking analysis was carried out to identify the most likely binding conformations and interaction affinity as scoring function of the original lead compound with respective target proteins. The binding affinities were calculated with generalized Born / volume integral (GB / VI) implicit solvent method implemented in MOE [15]. A refined docking step with energy minimization calculation was carried out using the top 30 poses of each molecule under the force field MMFF94x. Active sites present in the protein were identified from the 3D atomic coordinates of the receptor using Q-SITE FINDER [16]. It is an energy-based method for prediction receptor’s binding site, where a putative ligand may engender favorable interaction. The retrieved hits were docked into the binding site within the binding pockets of LEDGF-HIV-IN complex. All the atoms of the receptor molecule away from the ligand were kept rigid while receptor atoms in the locality of the ligand (in the binding site) were kept flexible and subjected to tether restraints to discourage gross movement. The ligand atoms were set free to move in the binding pocket. An energy minimization of LEDGF and HIV-IN proteins binding pockets was performed before binding affinity calculation for each hit. Different confirmations of the protein-ligand interactions were generated and the top ranked poses were selected. The binding affinity was calculated for each hit after energy minimization, and reported in unit of Kcal/Mol.
RESULTS AND DISCUSSION
Hotspot interactive residues and pharmacophore model generation
The complex crystal structure revealed that the Ile-365 residue of LEDGF IBD develop essential interaction with chain A residues, i.e. Thr-174 and Met178 and chain B residues, i.e. Leu-102,Ala-128, Ala-129, Trp-132 residues of IN. Whereas the Asp-366 residue of LEDGF-IBD interact with the IN chain A residues, i.e. Glu-170 and His-177 [17]. The structure elucidates the mode of recognition between these proteins and reveals a potential target site on IN for the designing small molecule inhibitors of the LEDGF–IN interaction. The MOE site finder analysis was performed after complex structure energy minimization to identify the nearby pocket able to anchor the lead hit and to check for the active site like potential target residues to be selected for pharmacophore model generation. The critical residues been identified during this analysis were Asp-167, Gln-168, Met-178, His-356, Lys-360, Ile-365, Asp-366, Phe-406 as potential active sites capable to hold small molecules. Several of these residues are identified as promising for biological interaction built-up between LEDGF and HIV-IN as discussed above. Therefore these residues of the LEDGF/p75-HIV-IN complex were selected as hotspot for structure-based pharmacophore model generation (Figure 1).
![[A] The 3D pharmacophore model within the complex of LEDGF/P75-HIV-IN indicating the exclusion volume spheres under default settings. [B] The 2D pharmacophore model of the hotspot residues of HIV-IN within the complex. The red dotted arrows indicate the hydrogen bond acceptor and the green dotted arrows indicate the hydrogen bond donor features. The yellow spheres representing the hydrophobic features in putative pharmacophore model.](https://www.jscimedcentral.com/public/assets/images/uploads/image-1706079317-1.PNG)
Figure 1: [A] The 3D pharmacophore model within the complex of LEDGF/P75-HIV-IN indicating the exclusion volume spheres under default settings. [B] The 2D pharmacophore model of the hotspot residues of HIV-IN within the complex. The red dotted arrows indicate the hydrogen bond acceptor and the green dotted arrows indicate the hydrogen bond donor features. The yellow spheres representing the hydrophobic features in putative pharmacophore model.
Pharmacophore based TIMBAL database screening
The validated pharmacophore-model was used to screen compounds with similar features from the TIMBAL database to find other novel drug-like compounds fulfill the specified criteria of the model. TIMBAL holds about 7,000 small drug-like molecules potent for protein-protein complexes [13]. Screening of entire TIMBAL repository through LigandScout identified single hit compound with a pharmacophore fit score of 105.66. Visual inspections showed that the hit compound exhibit selective interaction with both LEDGF and HIV-IN residues and seem to be promising for disruption of biological interactions within target complex. The lead compound following the Lipinski’s rule of five and hold six hydrogen bond acceptor and one hydrogen bond donor features (Figure 2).
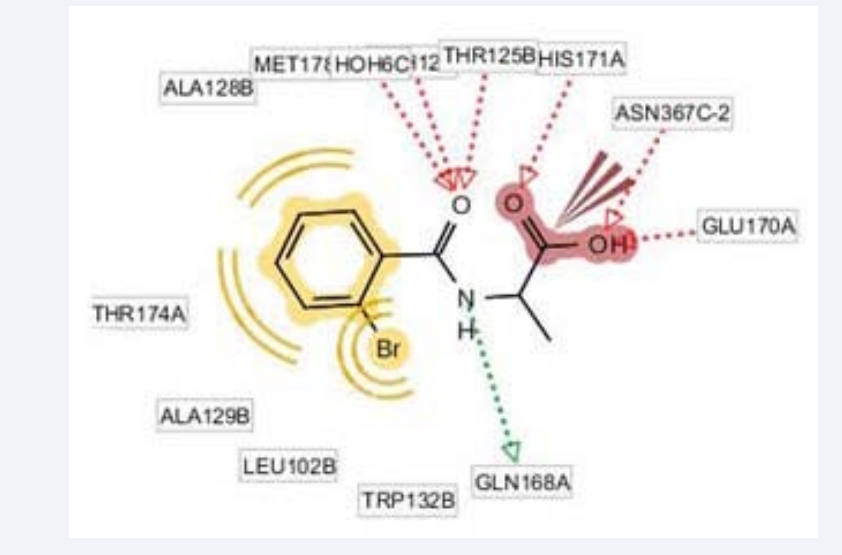
Figure 2: The 2D structure and IUPAC name of the lead compound from TIMBAL database, i.e. 2 (2-bromobenzamido) propanoic acid interaction sketch within target complex binding pocket. The lead compound develop interactions with key interacting residues of LEDGF–HIV-IN complex.
Molecular Docking
The MOE docking protocol used to search the binding modes of hit compound with the active site components accordingly. The lead hit was docked within the complex components. Besides TIMBAL lead compound some other source compounds previously reported as promising for the LEDGF-HIV target were also considered. These sources databases are; IPPI DB [18], KEGG DB [19] and HIP DB [20]. The KEGG & HIP DB sources hold one common compound. A lead compound from TIMBAL along with compounds from these sources are bring in same library and tested against target complex through molecular docking. The analyses inferred that the lead compound from TIMBAL database follow more or less same interaction features and docking score as according to other source compounds. (Figure 2; Table 1).
Table 1: The molecular docking score and drug-like features of TIMBAL library lead compound and other source anti-HIV IN inhibitor compounds.
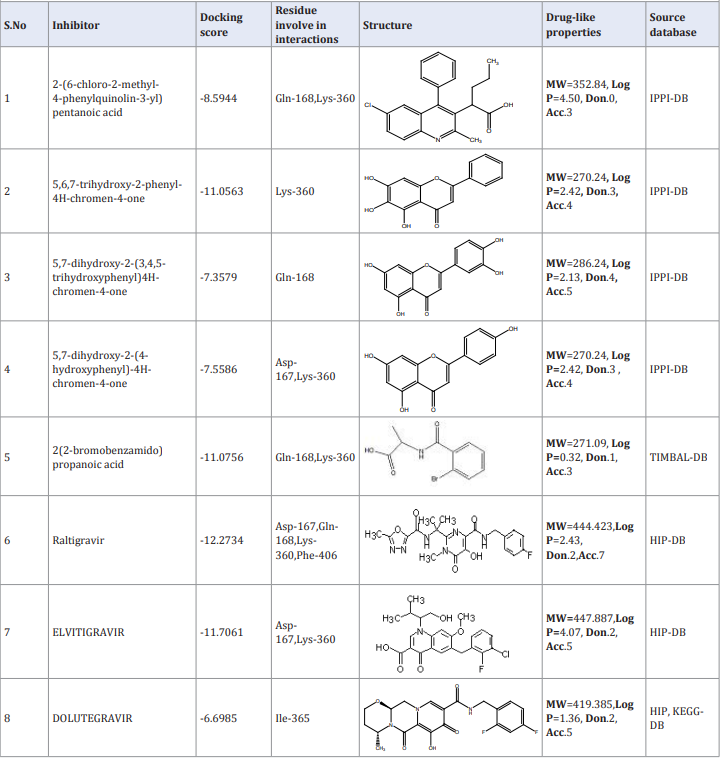
The docking results revealed that the TIMBAL hit is more effectively fitted into the LEDGF/P75-HIV-IN complex active site and target the hotspot region include the Asp-167, Gln-168 residues of IN and Lys-360 residue of LEDGF/p75. This speculates that the lead compound is capable to block biological interactions between LEDGF/P75-HIV-IN complex sub-units. Moreover, the comparative small molecular weight of hit compound from TIMBAL database make it more viable drug-like compounds compare to other source compounds. Therefore we assume this lead compound as best inhibitor to target the interaction between human LEDGF/P75 and HIV-IN and to design new anti-viral drugs.
CONCLUSION
The human host LEDGF/p75 and HIV-IN proteins biological interaction eventually lead to HIV infection. We employed the in silico drug discovery approaches to identify potential small molecule from TIMBAL source database capable to abolish these biological interactions between LEDGF/p75 and HIV-IN proteins. To the best of our knowledge, the TIMBAL repository never been screened so far against LEDGF/HIV-IN drugable sites. We found single lead compound from TIMBAL repository capable to perturb biological interaction between host LEDGF and pathogen HIV-IN proteins. The putative lead compound addressed in this study may provide a road map for identification of additional small molecules to be tested for the same purposes in future. The experimental evaluation of lead compound will be important for future anti-viral drug discovery. The lead chemical scaffold might be useful tool for better understanding the function and pathophysiological role of individual signalosomes, by disrupting the host LEDGF and pathogen HIV-IN binding interactions in the complex.









































































































































