In silico Toxicology - A Tool for Early Safety Evaluation of Drug
- 1. Sreekrishna college of Pharmacy and Research Centre, India
- 2. The Dale view college of Pharmacy and Research Centre, India
ABSTRACT
In silico toxicology plays a vital role in the assessment of safety/toxicity of chemicals and the drug development process. In silico toxicology aims to complement existing toxicity tests to predict toxicity, prioritize chemicals, guide toxicity tests, and minimize late?stage failures in drugs design. We provide a comprehensive overview, explain, and compare the strengths and weaknesses of the existing modeling methods and algorithms for toxicity prediction with a particular emphasis on computational tools that can implement these methods and refer to expert systems that deploy the prediction models.
KEYWORDS
• In silico toxicology
• Computer aided drug design
• Toxicity testing
• Recent advances
CITTATION
Dhanya S, Lal K, Reena SR (2018) In silico Toxicology - A Tool for Early Safety Evaluation of Drug. J Bioinform, Genomics, Proteomics 3(1): 1030
INTRODUCTION
Drug design, sometimes referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target [1]. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessary relies on computer modeling techniques [2]. This type of modeling is often referred to as computer aided drug design. Finally, drug design that relies on the knowledge of the three – dimensional structure of the biomolecular target is known as structure-based drug design.
The addition of computer-aided drug design (CADD) technologies to the research and drug discovery approaches could lead to a reduction of up to 50% in the cost of drug design .Designing a drug is the process of finding or creating a molecule which has a specific activity on a biological organisms. Development and drug discovery is a time-consuming, expensive and interdisciplinary process whereas scientific advancements during the past two decades have altered the way pharmaceutical research produces new bioactive molecules. Advances in computational techniques and hardware solutions have enabled in silico methods to speed up lead optimization and identification.
DRUG DISCOVERY PROCESS
The development of new drugs is very complex, costly and risky. The process of drug discovery involves a combination of many disciplines and interests starting from a simple process of identifying an active compound. The discovery of a new chemical entity that modifies a cell or tissue function is but the first step in the drug development process. Once shown to be effective and selective, a compound which is to be discovered must be completely free of toxicity, should have good bioavailability and marketable before it can be considered to be a therapeutic entity. The initial research, often occurring in academia, generates data to develop a hypothesis that the inhibition or activation of a protein or pathway will result in a therapeutic effect in a disease state. The outcome of this activity is the selection of a target which may require further validation prior to progression into the lead discovery phase in order to justify a drug discovery effort. During lead discovery, an intensive search ensues to find a drug-like small molecule or biological therapeutic, typically termed a development candidate, that will progress into preclinical, and if successful, into clinical development and ultimately be a marketed medicine (Figure 1).
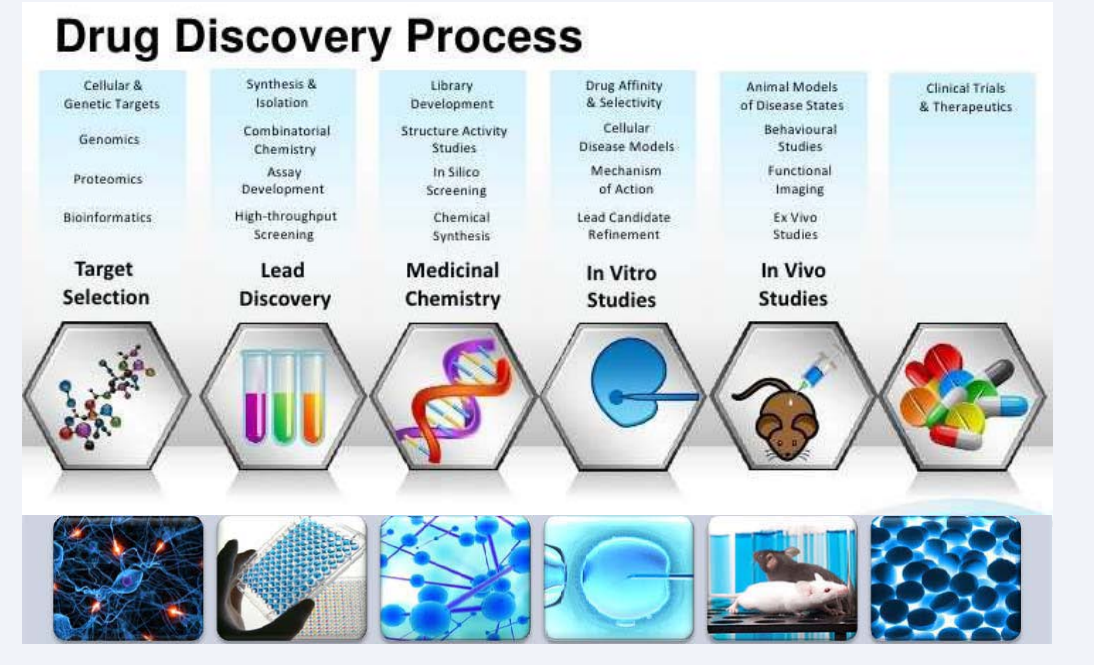
Figure 1: Schematic representation of drug discovery process.
In silico DRUG DESIGN
Computers are an essential tool in modern medicinal chemistry and are important in both drug discovery and development. Rapid advances in computer hardware and software have meant that many of the operations which were once the exclusive province of expert can now be carried out on ordinary laboratory computers with little specialist expertise in the molecular or quantum mechanics involved. The advent of software in the world of drug discovery has enabled the development of novel drug candidates as a more sophisticated, precise and rapid process. Computers can be used to simulate a chemical compound and design chemical structures. Computer-aided design including quantitative energy calculations and graphical methods has been rapidly introduced in the pharmaceutical industry. Potential compounds are modeled computationally to estimate their ‘fit’ to the target by computing a scoring function or an energy function. Most algorithms consider both structural and functional interactions such as steric fit, hydrogen bonding and hydrophobic interactions. Thus, a computer can show scientists what the receptor site looks like and how one might tailor a compound to block an enzyme from attaching there.
Advantages of in silico methods
- Reduce costs
- Reduce time to market
- Reduce side effects
- Improve success rate
- Improve bioavailability & bioactivity
- Improve understanding of drug-receptor interactions
- Improve understanding of molecular recognition process
- Less man power is required
In silico TOXICOLOGY
It is a vibrant and rapidly developing discipline that integrates information and data from a variety of sources to develop mathematical and computer based models to better understand and predict adverse health effects caused by chemicals such as environmental pollutants and pharmaceuticals. Toxicity is a measure of any undesirable or adverse effect of chemicals. Toxicity tests aim to identify harmful effects caused by substances on humans, animals, plants, or the environment through acute exposure (single dose) or multiple exposure (multiple doses). Several factors determine the toxicity of chemicals, such as route of exposure (e.g., oral, dermal, inhalation), dose (amount of the chemical), frequency of exposure (e.g., single versus multiple exposure), duration of exposure, ADME properties (absorption, distribution, metabolism, and excretion/ elimination), biological properties (e.g., age, gender), and chemical properties. Animal models have been used for a long time for toxicity testing. However, in vitro toxicity tests became plausible due to the advances in high throughput screening. In silico toxicology (computational toxicology) is one type of toxicity assessment that uses computational resources (i.e., methods, algorithms, software, data, etc.) to organize, analyze, model, simulate, visualize, or predict toxicity of chemicals. It is intertwined with in silico pharmacology, which uses information from computational tools to analyze beneficial or adverse effects of drugs for therapeutic purposes [2].
Computational methods aim to complement in vitro and in vivo toxicity tests to potentially minimize the need for animal testing, reduce the cost and time of toxicity tests, and improve toxicity prediction and safety assessment. In addition, computational methods have a unique advantage of being able to estimate chemicals for toxicity even before they are synthesized. In silico toxicology encompasses a wide variety of computational tools (A) databases for storing data about chemicals, their toxicity, and chemical properties; (B) software for generating molecular descriptors; (C) simulation tools for systems biology and molecular dynamics; (D) modeling methods for toxicity prediction; (E) modeling tools such as statistical packages and software for generating prediction models; (F) expert systems that include pre-built models in web servers or standalone applications for predicting toxicity; and (G) visualization tools.
TOXICITY PREDICTION OF A MOLECULE
Molecule is formed when two or more atoms combine together chemically (e.g.: H2 O). A compound is said to be a molecule when it contains at least two different elements (e.g.: H2 O). All compounds can be called as molecule but all molecules are not compounds. In terms of pharmacology or in biochemistry, a small molecule is an organic compound which has low molecular weight and may act as a substrate or inhibitor. In medical field, the term is restricted to the molecule that binds to a biopolymer and act as an effector. Most of the small molecules are drug molecules.
Drug molecules are potential lead molecules which act as therapeutic agents and gives beneficiary effects. To come up with single potential lead molecule it takes 12 -16 years. Besides the beneficiary aspects, there may be adverse effects also when using drug/potential lead molecules.
It has been known that, most well known drugs are poisonous substances. All useful drugs produce unwanted effects due to complex nature of human body. Some drugs are more adverse and can produce dangerous effects. So, toxicity is more important measurement during the synthesis of a molecule. One knows that it is difficult to synthesize a potential lead molecule in a shorter time period by undergoing all types of tests.
Computer Aided Drug Designing approaches to design a drug molecule using different tools to predict the pharmacokinetic properties (what the body does to the drug when the drug is administered). The pharmacokinetic properties are also stated as ADME-Tox (Absorption, Distribution, Metabolism, Distribution and Toxicity).
Drug failures due to toxicity can only be known in the later stages of clinical trials. To minimize the time required by these clinical trials, determination of toxicity potential as early as possible using Insilco prediction is very essential. With the richness of combinatorial library and high throughput screening, prediction on drug toxicity is easier and possible even before the synthesis of the molecule.
(* Insilco – expression used to mean “performed on computer or via computer simulation”)
Synthesizing a single new drug molecule typically takes 12 -16 years and in most of the cases these molecules are rejected because of failure in clinical trials at the level of toxicity. Pharmaceutical companies have recently come up with ADME and toxicity test with the help of IN SILICO based approaches. These approaches can be used to predict the toxicity of a drug molecule even before its synthesis. Even though the IN SILICO approaches are quiet easier, there are problems to overcome.
- Toxicity may refer to a wide range of effects like carcinogenicity, cytotoxicity.
- There is insufficient data, particularly in the case of humans.
- The Insilco methods are class specific, determining whether toxicity is on or off are least accurate.
Drug molecules can cause toxicity in many ways, like it may not be the drug itself that causes the toxicity, the metabolite may also cause some unwanted effects. In some cases, the cytotoxic and mutagenic properties of the drug molecules are selected to kill the diseased or cancer cells but it has a high probability that it may also affect normal cells.
Toxicity measurement
Toxicity is a quantity that can be measured; the simple measure of toxicity is LD50. It is a drug dose which kills 50% of treated animals within a period of time. The therapeutic window gives the range of the dosage between the minimum effective therapeutic concentration and the minimum toxic concentration.
There are many tools to predict the toxicity of a molecule, some of them are commercial, some are online web servers and few of them are freely downloadable (Figure 2).
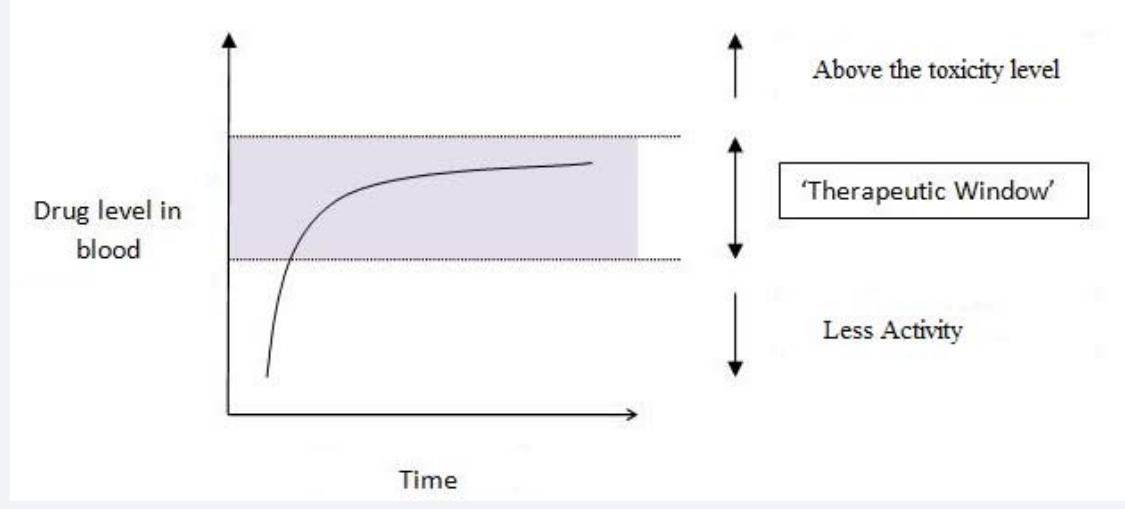
Figure 2: Gr aph of Therapeutic window.
SOFTWARES TOOLS USED IN IN SILICO TOXICOLOGY
There are a host of statistical expert systems though TOPKAT, PASS, MCASE, TEST, CAESAR, LAZAR & OCHEM are most commonly known.
TOPKAT (Toxicity Prediction by Komputer Assisted Technology)
It contains a number of models covering a range of different toxicity endpoints (http://accelrys.com/products/collaborative-science/biovia-discovery-studio/qsaradmet-and-predictive-toxicology.html) including rat chronic LOAEL, skin irritation, eye irritation, developmental toxicity, rodent carcinogenicity, rat maximum tolerated dose, carcinogenicity potency TD50, acute oral rat, skin sensitization and Ames mutagenicity [3].
Many of the models provide a binary summary prediction whereas models such as those for acute oral toxicity estimate a LD50 value. A prediction is accompanied with information of whether a target substance of interest lies within the applicability domain of the model and associated confidence limits. A search for similar analogs within the training sets underpinning each model can be performed which provides an idea of the chemical coverage of the model for the target substance.
PASS (Prediction of Activity Spectra for Substances)
It is a commercial expert system which estimates the probability of over 4000 kinds of biological activity including pharmacological effects (e.g., antiarrhythmic), biochemical mechanisms (e.g., cyclooxygenase 1 inhibitor), toxicity (e.g., carcinogenic), metabolism (e.g., CYP3A4 inhibition), gene expression regulation (e.g., VEGF expression inhibition), and transporter related activities (e.g., P-glycoprotein substrate). Predictions are based on the analysis of structure activity-relationships for more than 250,000 biologically active substances including drugs, drug-candidates, leads and toxic compounds. A free version known as PASS Online (http://www.pharmaexpert. ru/passonline/) is available for use upon registration which allows predictions to be made on a substance by substance basis.
MCASE from multicase.com
It contains a wealth of different models for toxicity prediction (http://www.multicase.com/caseultramodels#model_bundle_ list). Endpoints include reproductive toxicity, developmental toxicity, renal toxicity, hepatotoxicity Ames mutagenicity, cardiotoxicity, and skin and eye toxicity amongst others. CASE Ultra is the end-user application of MCASE. The QSAR models are derived on the basis of biophores and the modulators. The program performs a hierarchical statistical analysis of a training set to uncover substructures that appear mostly in active chemicals therefore having a high probability for the observed activity – these are termed biophores. For each set of chemicals containing a specific biophore, the program identifies additional parameters called modulators, which can be used to derive QSARs within the reduced set of congeneric chemicals. The modulators consist of certain substructures or physicochemical parameters that significantly enhance or diminish the activity attributable to the biophore. QSARs are derived by incorporating both biophores and modulators into the model.
TEST (Toxicity Estimation Software)
It is a freely available software tool that houses a number of models that have been developed using a number of machine learning approaches by the US EPA. A user decides on an endpoint to be predicted based on a particular modeling approach. The prediction is reported along with a consensus prediction based on all 6 other approaches as appropriate as well as the performance of similar analogs from the training and test sets. There are three mammalian toxicity models available; namely rat oral LD50, Ames mutagenicity and developmental toxicity. The developmental toxicity model was developed under the auspices of the EU project CAESAR and implemented into TEST as an additional toxicity module.
CAESAR was an EU funded project which resulted in the derivation of a number of models for different endpoints including skin sensitization, Ames mutagenicity, carcinogenicity and developmental toxicity.
LAZAR (Lazy Structure-Activity Relationships)
It uses an automated read-across procedure to make predictions for a number of different endpoints. The approach is built onto the OpenTox (http://www.opentox.org/) framework within the ToxPredict application. LAZAR houses models for carcinogenicity in different species, Ames mutagenicity as well as maximum repeated dose (http://lazar.in-silico.de/predict).
OCHEM (Online Chemical Modeling Environment)
It is a web-based platform (http://www.ochem.eu) aimed at trying to simplify the steps needed to derive new QSAR models. The platform houses a database of experimental measurements and a modeling framework to allow users to derive new QSARs using the statistical algorithms from the available experimental data or to upload their own data. Users can register to access the system and the available QSAR models, elevated privileges are given to users who pay a fee or contribute in kind. To date the only toxicity model that is currently publically available is an Ames mutagenicity QSAR model. A set of over 1700 structural alerts (known as ToxAlerts) for a wide range of endpoints including genotoxicity, developmental toxicity and skin sensitization are also available.
TIMES (Tissue Metabolism Simulator)
It is nominally categorized as a hybrid expert system since it contains alerts some of which are underpinned by 3D QSARs. It enables predictions to be derived from local models whilst retaining the breadth of coverage for a wide range of chemicals. The strength of the TIMES platform lies in the fact that its models incorporate metabolism – there are models that predict skin sensitization potency, the outcome of an Ames mutagenicity test as well as the outcomes for in vitro chromosomal aberration and in vivo micronucleus tests. These models rely on both structure-activity and structure metabolism rules to make their predictions.
PreADMET
PreADMET is one of the online servers to predict ADME, toxicity, Drug likeness and molecular descriptor calculation. PreADMET predicts the mutagencity and carcinogencity of compounds, so that toxicity is avoided in compounds.
The input compounds given to PreADMET server is either by drawing the molecule or by uploading the “mol” format of that compound which is to be predicted. The compounds structure or the “mol” format can be obtained from different chemical databases like drug bank, Chembl, Pubchem, etc.
PreADMET tools use the strategy to obtain the model which can be used to predict absorption, distribution and toxicity.
In silico MODELING METHODS
Many in silico methods have been developed to predict the toxicity of chemicals. The methods we discuss here are chosen either because they illustrate the historical development of in silico toxicology or they represent the state-of-the-art method for predicting toxicity. For each method, we provide (if applicable) a mathematical description, discussion of strengths and limitations, recommendations about when and why to use the method, and existing tools that implement the method. Additionally, for the sake of clarity, we keep equations and visual representations of models as general as possible (Figure 3).
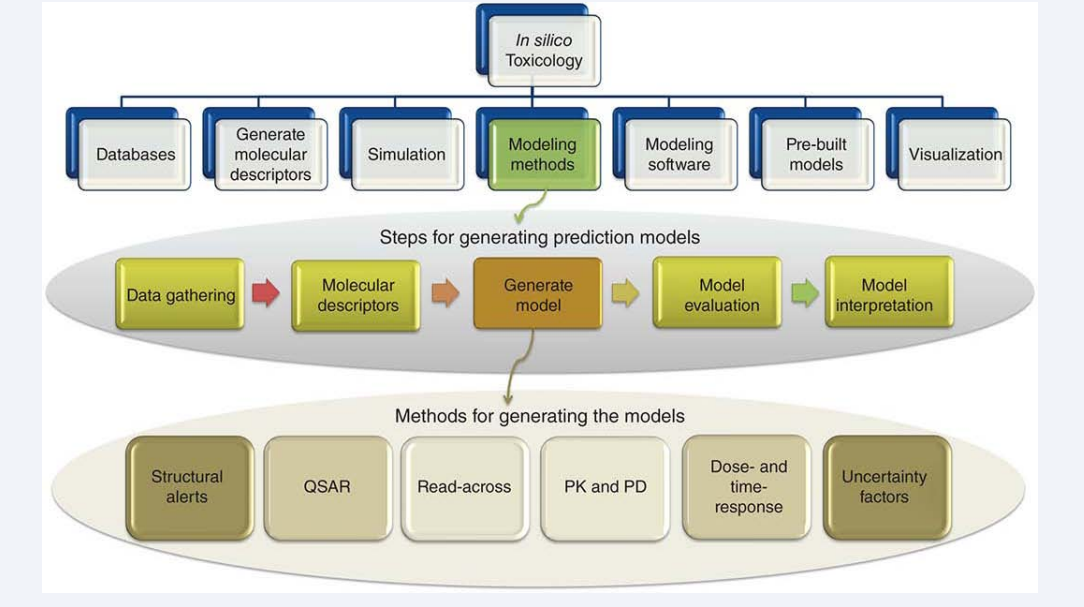
Figure 3: In silico toxicology tools, steps to generate prediction models, and categories of prediction models Structural Alerts and Rule-based Models.
Structural alerts (SAs) (also called toxicophores/toxic fragments) are chemical structures that indicate or associate to toxicity. SAs can consist of only one atom or several connected atoms. A combination of SAs may contribute to toxicity more than a single SA.
There are two main types of rule-based models that we will consider: human-based rules (HBRs) and induction-based rules (IBRs). HBRs are derived from human knowledge of field experts or from literature, but IBRs are derived computationally. HBRs are more accurate but are limited to human knowledge that could be incomplete or biased. Moreover, updating HBRs is often impractical as it requires detailed literature analysis. On the contrary, IBRs can be generated efficiently from large datasets. IBRs may propose hypotheses about associations between chemical structural properties (or their combinations) and toxicity endpoints, which may not be identified through human insights. IBRs are implemented using probabilities to determine if SAs correspond to the toxic or non-toxic class. It is possible to have hybrid-based rules systems that contain IBRs and HBRs, with new rules being generated computationally
It is easy to interpret and implement SAs. They are useful in drug design to determine how drugs should be altered to reduce their toxicity. Using structure to predict toxicity allows identifying the structure of potential metabolites.
LIMITATIONS OF STRUCTURAL ALERTS
- SAs use only binary features (e.g., chemical structures are either present or absent) and only qualitative endpoints (e.g., carcinogenic or non-carcinogenic).
- SAs do not provide insights into the biological pathways of toxicity and may not be sufficient for predicting toxicity.
- Depending on the concurrent absence or presence of other chemical properties, toxicity may decrease or increase.
- The list of SAs and rules may be incomplete, which may cause a large number of false negatives (i.e., toxic chemicals predicted as non-toxic) in predictions.
It is necessary to understand how to interpret the output of SA models. If a chemical does not include SAs or does not match any toxicity rules, this does not indicate non-toxicity. However, there should be a balance between the list of SAs and rules, their comprehensiveness, and predictive power. If SAs and rules are diverse, they can be applied to a large number of chemicals, but this may increase false positives (i.e., non-toxic chemicals predicted as toxic). However, if they are too narrow, they can be applied only to a small group of chemicals, and this may increase false negatives (i.e., toxic chemicals predicted as non-toxic).
An example of SA list for skin sensitization was published in 1982 by Dupuis and Benezra. Another SA list was proposed by Ashby and Tennant in 1988 to predict carcinogenicity and mutagenicity, hepatotoxicity, cytotoxicity, irritation/corrosion of skin, and eye and skin sensitization.
CHEMICAL CATEGORY, READ-ACROSS, AND TREND ANALYSIS
A chemical category is a group of chemicals whose properties and toxicity effects are similar or follow a similar pattern. Chemicals in the category are also called source chemicals. The OECD Guidance On Grouping Of Chemicals lists several methods for grouping, such as chemical identity and composition, physicochemical and ADME properties, mechanism of action (MoA), and chemical/biological interactions.
Read-across is a method of predicting unknown toxicity of a chemical using similar chemicals (called chemical analogs) with known toxicity from the same chemical category.
Trend analysis is a method of predicting toxicity of a chemical by analysing toxicity trends (increase, decrease, or constant) of tested chemicals. A hypothetical example of trend analysis shows that when carbon chain length (CCL) increases, acute aquatic toxicity increases.
A summary of different parameters that must be considered when designing a read-across model is depicted in Figure (4).
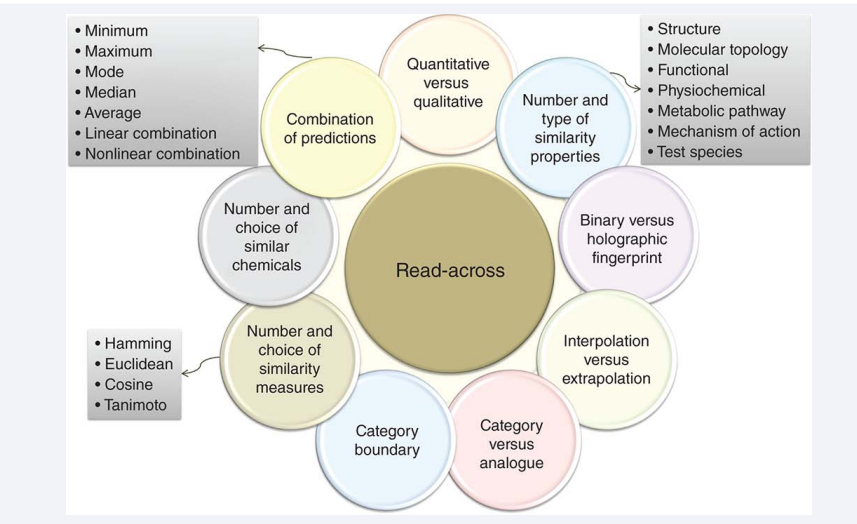
Figure 4: Different properties of read-across models.
There are two ways to develop a read-across method: analog approach (AN) (called one-to-one), which uses one or few analogs, and a category approach (CA) (called many-to-one), which uses many analogs. AN may be sensitive to outliers because two analogs may have different toxicity profiles. Using many analogs for CA is useful to detect trends within a category and may increase confidence in the toxicity predictions. Identifying similar chemicals can be done in two steps: representing chemicals as feature vectors of chemical properties, and then calculating similarity of chemicals. The first step is implemented using either binary or holographic fingerprints. A binary fingerprint is a feature vector of binary bits representing presence (1) or absence (0) of a property (e.g. presence of a methyl group). However, a holographic fingerprint uses frequency of properties (e.g. number of methyl groups). Continuous chemical properties (e.g., melting point) can be used as well.
Advantages of read across
- Read-across is transparent
- Easy to interpret and implement
- Read-across can model quantitative and qualitative toxicity endpoints, and it allows for a wide range of types of descriptors and similarity measures to be used to express similarity between chemicals.
Limitations of read across
- Statistical similarity measures do not provide biological insight of toxicity.
- complex similarity measures may complicate model interpretation.
- In reality, read-across uses small datasets compared to other approaches such as QSAR because there are usually only a few analogs for a given chemical.
Read-across was applied to predict: - carcinogenicity, hepatoxicity, aquatic toxicity, reproductive toxicity, skin sensitization, and environmental toxicity.
Examples of tools implementing read-across are The OECD QSAR Toolbox, Toxmatch, ToxTree, AM-BIT, AmbitDiscovery,AIM, DSSTox, or ChemIDplus. .
Dose–Response and Time–Response Models
Dose–response (or time–response) models are relationships between doses (or time) and the incidence of a defined biological effect (e.g., toxicity or mortality). A dose is ‘the total quantity of a substance administered to, taken up, or absorbed by an organism, organ, or tissue and can be measured with in vitro or in vivo experiments.’ Time can be the time to produce a response or the time for recovery.
Exposure time can be continuous, intermittent, or random, and exposure can be acute, short-term, sub-chronic, and chronic exposure. Time–dose models describe the relationship between time and dose for a constant response.
Limitations are
- The three models cannot extrapolate to other chemicals.
- Time–response models cannot extrapolate to other doses of the same chemical.
- Time–response models require that tested individuals have uniform susceptibility levels, or these models may be unreliable if some individuals have an extremely low or high resistance.
- If time intervals are long, time–response models may overestimate or underestimate the response at a given moment.
These time- response and dose-response models are complementary to one another and must be used together to achieve reliable conclusions.
Several databases include CEBS, PubChem .These models were used for modeling rectal cancer, mutagenicity and developmental toxicity.
PHARMACOKINETIC MODELS
Pharmacokinetic (PK) models relate chemical concentration in tissues to time, estimate the amount of chemicals in different parts of the body, and quantify ADME processes. Toxicokinetic models are PK models used to relate chemical concentration in tissues to the time of toxic responses. PK models can be compartmental and non-compartmental. A compartment is the whole or part of an organism in which the concentration is uniform. Compartmental models consist of one or more compartments, and each compartment is usually represented by differential equations.
One-compartment models represent the whole body as a single compartment, assume rapid equilibrium of chemical concentration within the body after administration, and do not consider the time to distribute the chemical. However, these models do not consider the distribution time of chemicals. Two-compartment models consist of two compartments: central (for rapidly-perfused tissues e.g., liver or kidney) and peripheral (for slowly perfused tissues e.g., muscle or skin).
On the other hand, physiologically based pharmacokinetic (PBPK) models include, in addition to concentration and time, physiological descriptors of tissues and ADME processes such as volumes, blood flows, chemical binding/partitioning, metabolisms, or excretions.
PHARMACODYNAMIC MODELS
Pharmacodynamic (PD) models relate a biological response to the concentration of chemical in tissue. Toxicodynamic models are PD models that relate toxicity to the concentration of the chemical. PD models that are based on anatomy, physiology, biochemistry, and biology are called physiologically based pharmacodynamic (PBPD) models.
Examples of PK and PD modelling tools are WinNonlin, Kinetica, and ADAPT 5. For example, PBPK was used for route-to-route extrapolation,toxicity and risk assessment, and carcinogenicity assessment.
Recent Advances in In Silico Toxicology
Nano toxicity:
Nano toxicity is the study of adverse effects caused by nanomaterials. Nanomaterial’s are small particles on the nanoscale (10−9 m) size range. When a particle size is decreased within the nanoscale size range, its physical and chemical properties are changed, affecting its toxicity. It was found that nanoparticles cause different or worse toxicity effects than the larger particles of the same substance. A nanoparticle can be toxic even if the particle is not toxic at a larger size. The small size of nanomaterials facilitates cell membrane penetration and biodistribution.
Properties affecting toxicity of nanomaterials:
- The shape of nanomaterials affects toxicological responses. For example, isolated long fiber carbon nanotubes are more inflammogenic in the outer regions of the lung than non-fibrous nanotubes.
- Large surface areas of nanomaterials increase the contact area with the biological environment and their chemical reactivity.
- Surface coating material of the nanomaterials can affect biological functions. It was found that toxicity of nanomaterials that have the same metallic core could be predicted by using the properties of the coating material..
- Other physicochemical properties such as electrostatic interactions between nanomaterials and biological targets can influence toxicity.
Mechanisms of nanoparticles inducing toxicity:
- Interaction and binding of the nanoparticle’s surface with a biological environment (e.g., protein or cells)
- Cellular entry: nanoparticles potential to enter cells
- Release of ions from the surface: ionic forms of metals can be more active and
- Generation of reactive oxygen species (ROS): overproduction of ROS can cause oxidative stress and inflammation, which disrupt normal biological functions and damage DNA and proteins (Figure 5).
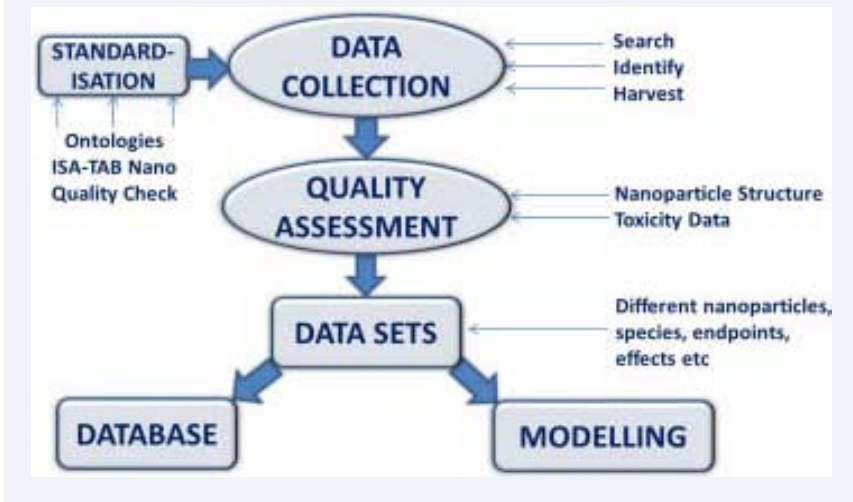
Figure 5: Schematic representation of analyzing methods.
TOXICITY TEST
In order to ensure drug safety a variety of toxicological evaluations are performed, to the requirement of national regulatory authorities such as the Food and Drug Administration (FDA) in the United States and the European Medicines Evaluation Agency (EMEA) in the European Union [4]. The tests that may be performed are:
- Acute toxicity (24 h)
- Prolonged and chronic toxicity (30-90 days)
- Mutagenicity
- Carcinogenicity
- Teratogenicity
- Reproductive effects
- Skin sensitization (including photosensitization)
- Skin irritation
- Eye irritation
- Immunological assessment.
Acute toxicity
Acute toxicity tests are normally performed in rats and mice and occasionally in the rabbit and guinea pig. The purpose of the assay is to determine ultimately the LD50, or the dose of drug that will be lethal to 50% of a population. The time period of the test is short (normally 24 h and occasionally up to 48 h) with observation following dosing of up to 14 days.
Dosing the animals may be via a number of routes including intravenous, intraperitoneal, dermal, oral and inhalation. Organization for Economic Co-operation and Development (OECD) guidelines provide experimental details for oral (OECD Guideline 401) and dermal dosing (OECD Guideline 402) as well as the inhalation route (OECD Guideline 403; details of the OECD guidelines can be obtained from their internet site. Whilst the acute toxicity test is relatively simple to perform, it can provide the experienced toxicologist with a wealth of information far beyond the basic LD50, including information on observational and physiological effects. The LD50 test is now not acceptable in many countries and has been replaced by the fixed dose, up-and-down, and acute toxic class procedures.
Repeated dose toxicity assays
Toxicological assessment for longer time periods is required as a drug progresses through the development process. This provides toxicological information regarding exposure to drugs, normally at sub-lethal concentrations, over a more realist timeframe. Short-term repeated dose studies (OECD Guideline 407) last between 14 and 28 days. Dosing is graded in 3-4 concentrations with the highest dose designed to cause some toxicity, but not lethality. Normally between 5-10 rats of each sex (though mice and dogs are also utilized) are tested per group. At the completion of the test a whole host of clinical and histological evaluations are recorded, including experimental observations and whole body and individual organ analysis. Such information will clearly enhance that gathered from acute toxicity studies. Other subchronic toxicity studies are maintained for up to 90 days (OECD Guideline 408). Again animals are exposed to the drug continuously and potentially via a number of different routes. This provides much information regarding organ toxicity.
Testing for carcinogenicity
Carcinogenicity assays may be considered as an extension of the chronic toxicity test. To test a pharmaceutical substance for carcinogenicity is a lengthy and expensive process. Typically, a substance is tested in two species (rats and mice) and both sexes with continuous exposure for up to 24 months. Exposure is typically via the oral route.
Tests for reproductive toxicity and teratogenicity
The determination of the effects of chemicals on the reproductive ability of males and females, as well as issues such as teratogenicity, is a broad and complex area. It is generally accepted that teratogenicity is only a part of potential reproductive toxicity. Testing for teratogenicity (abnormal foetal development) requires the in utero exposure of the foetus to a drug. Tests are normally performed in the rat, although the rabbit may occasionally be used.
Typically three dose levels are applied, the highest being that which will induce some limited maternal toxicity; the lowest dose will cause no maternal toxicity. For rat tests 20 pregnant females will be used at each dose level. The drug is administered during the development of the major organs in the foetus (e.g., 6-15 days after conception). The animals are dosed via their drinking water. The test is terminated on the day prior to normal delivery and foetal development, in terms of both the number of live foetuses, and of any malformations present, is assessed. For the more accurate assessment of effects of a drug on species fecundity and between generations, a multigeneration toxicity test is required. A good example is the two-generation reproduction toxicity test (OEeD Guideline 416), although these tests may also be maintained for three or more generations. Such assays are performed usually with rats at three dose levels. The highest dose level is typically one-tenth of the LD50; the lowest should cause no sub-chronic toxicity. Males and females are treated with the drug for 60 days, after which they are allowed to mate. Subsequent generations are assessed for a wide variety of endpoints including: number of live births; abnormalities at birth; gender and weight at birth; histological examinations, etc.
Alternatives to toxicity testing
It can be concluded from this brief review of toxicological methods that the experimental assessment of drug toxicity is a time-consuming, expensive, yet essential, part of the pharmaceutical research and development process. The consequence of a drug being found to be toxic at a late stage of development could be immensely costly to a company. With this in mind, methods are constantly being sought to determine drug toxicity as early and as cheaply as possible. Much effort has been placed into the development of in vitro assays, cell culture techniques and most recently DNA arrays as replacements for toxicity testing. In addition, a number of computer-aided toxicity prediction methods are available. These are based on the fundamental premise that the toxicological activity of a drug will be a function of the physico-chemical and/or structural properties of the substance. Once such a relationship has been established, further chemicals with similar properties can be predicted to be toxic.
COMPUTER AIDED PREDICTION OF TOXICITY AND METABOLISMS
The development of computer-aided toxicity and metabolism prediction techniques can be broadly classed into three areas:
- Quantitative structure-activity relationships (QSARs)
- Expert systems based on QSARs
- Expert systems based on existing knowledge.
Quantitative structure-activity relationships
QSARs attempt to relate statistically the biological activity of a series of chemicals to their physico-chemical and structural properties. They have been used successfully in the lead optimization of drug and pesticide compounds for over three decades. They have also been applied to the prediction of toxicity. The most straightforward QSARs have been developed for acute toxicity, with relatively restricted groups of compounds, about which something of the mechanism of action is known.
In this approach the toxicity of the compounds is described as a function of their ability to penetrate to the site of action, or accumulate in cell membranes (a hydrophobic phenomenon parameterized by log P), and their ability to react covalently with macromolecules (an electrophilic phenomenon parameterized by ELUMO). A number of caveats to this model are immediately obvious. Firstly, whilst this model clearly fits the data well, it does so only for a relatively small number of structurally similar molecules. Its application to predict drug toxicity is likely to be extremely limited. It is envisaged that for the efficient prediction of acute toxicity a tiered approach combining both structural and physico-chemical rules with such QSARs will be required. Such rules may direct the prediction to be made from, for example, separate QSARs for aliphatic and aromatic molecules, or account for effects such as ionization or steric hindrance of reactive centers. It should be noted that such a tiered approach still requires more effort in the measurement of toxicological activity and modeling.
The second caveat relates to the nature of the biological activity itself. It should be no surprise that the data have been obtained from an in vitro toxicological assay. Such data are quickly and cheaply determined (and can be obtained in one laboratory, as in this case). Undoubtedly for drug toxicity a model based on mammalian LD50 data would be preferable. However, very few such QSARs are available. The reason for this is believed to be that there are not sufficient “quality” toxicity data on which to develop these QSARs.
Whilst a large number of toxicity data may be available on databases such as the Registry of Toxic Effects of Chemical Substances there is no consistency in the data, and many attempts to model such data are clearly hindered by the excessive error and inter-laboratory variation that may exist. Johnson and Jurs [5] report a predictive model for the acute oral mammalian toxicity for 115 anilines using data retrieved from the RTECS database. The model utilizes a neural network based on five physico-chemical descriptors. It is reported to predict the toxicity of an external validation set well. Further unpublished analysis of the toxicity data suggests that there may, however, be considerable variation in the data, following a comparison of the toxicity data for positional isomers in the data set. Other workers have investigated the acute toxicity data from RTECS to rats and mice (based on an arbitrary categoric scale) to develop a model based on a decision tree approach for aliphatic alcohols. QSARs have been developed for a whole range of other toxicity endpoints, especially those that provide a quantitative determination of toxicity.
Expert systems for toxicity prediction
“Expert system” is taken to mean a computer-assisted approach to predict toxicity. Expert systems based on QSARs The logical extension of the QSAR approach to make large scale predictions of toxicity is to computerize it. A simple DOS-based QSAR program is MicroQSAR. Following the entry of a SMILES string a wide variety of endpoints are predicted. Whilst most of these are environmental in nature, a number of human health effects are also predicted. Since its inception, the program has not however been developed to achieve its full potential. Another environmentally-based prediction program is ECOSAR. This operates by assigning a molecule to a particular class to make a prediction (normally based on log P), and has, as such, been criticized for the arbitrary manner in which classes are identified. As such, it gives the user the opportunity to see the method (and view the result) of the prediction that will be made by the United States Environmental Protection Agency (U.S. E.P.A.). TOPKAT, and the related program Q-TOX, are probably the best known and commercially successful QSAR based expert system prediction programs. TOPKAT was developed by Health Designs Inc., a wholly owned subsidiary of Accelrys. The link up with Accelrys means that the TOPKAT system is now integrated with a number of other tools, such as the TSAR molecular spreadsheet. The power of this is clearly the ability to make predictions from the spreadsheet. TOPKAT makes predictions for a number of toxicity endpoints, including: carcinogenicity; mutagenicity; developmental toxicity; maximum tolerated dose; various acute toxicities and others.
An appreciation of the ethos behind the toxicity prediction systems is important to understand their capabilities. TOPKAT models are developed from large heterogeneous databases of compounds, normally obtained from sources such as the RTECS database, and the open literature. The toxicity of these compounds may thus be measured by a variety of different methods in a number of different laboratories. Relationships are sought between the toxicity and any of 1000s of different physico-chemical and structural indices. These indices are normally based on topological and electrotopological properties of the whole molecules and individual atoms within them. It is thus often difficult to assign any mechanistic meaning and thus confidence to these models. A particular strength of the TOPKAT system is that it provides an estimate of confidence that can be attached to the prediction. The so-called “Optimum Prediction Space” will indicate whether the prediction has been made from information (in terms of the training set of molecules) similar to that of the predicted molecule.
Another well recognized prediction methodology is the computer automated structure evaluation (CASE) technique. This was developed by Klopman and coworkers and the CASE technique drives a number of systems including CASETOX, CASE, MULTICASE and TOXALERT [6]. Predictive models have been developed for a number of toxicological endpoints including carcinogenicity; mutagenicity; teratogenicity; acute toxicities as well as physico-chemical properties. The CASE models are derived from large and heterogeneous data sets. Compounds are split into fragments ranging from two to n atoms (though fragments greater than eight atoms are likely to be unwieldy). The fragments are then assessed statistically to determine if they may promote the biological activity (biophores) or decrease it (biophobes). Once fragments have been identified, they can be used either as “descriptors” in a regression-type approach to predict toxicity, or occasionally as structural alerts for rule-based systems. As with TOPKAT, this approach requires large sets of toxicological data, which will inevitably include compilations from the open literature. Both techniques do, however, provide predictive models from such data in a short period of time. The CASE approach lacks a mechanistic approach to identify the fragments (it is simply a statistical analysis), although it is suggested that mechanistic interpretation of the fragments can be applied a posteriori. Recently the United States Food and Drug Administration have come to a Cooperative Research and Development Agreement (CRADA) with Multicase Inc. to develop the carcinogenicity model, with the inclusion of proprietary regulatory data.
Expert systems based on existing knowledge
There are a number of expert systems that make predictions of toxicity from a “rule-based” approach. These are expert systems in their purest form, which capture the knowledge of an expert for utilization by a non-expert.
The power and utility of these systems is reliant upon two items: firstly adequate software is required to comprehend and interpret chemical structures; and secondly knowledge is needed to form the rule-base of the expert systems. The former is well developed and a number of software packages are commercially available as detailed herein; the latter, for many toxicological endpoints, is still at a rudimentary level. The software packages developed by LHASA Ltd provide a good illustration of the systems available to predict toxicological and metabolic endpoints. LHASA Ltd. itself is a unique company amongst the expert system providers. LHASA Ltd. has charitable status and is the coordinator for a collaborative group of “customers” who purchase its products (in particular DEREK (Deductive Estimation of Risk from Existing Knowledge) for Windows™).
The collaborative group includes members from the pharmaceutical, agrochemical, and personal product industries, as well as regulatory agencies, from Europe and North America. At the time of writing there are over 20 members in the collaborative group. Members of the group are given the opportunity to contribute their own knowledge to the development of new rules. Probably the most developed product from LHASA is the DEREK for Windows™ software, which provides qualitative predictions of toxicity from its rule base. As with all such systems, the concept is simple: namely that if a particular molecular fragment is known to cause toxicity in one compound, if it is found in another compound the same toxicity will be observed. The system is driven by the LHASA software originally written in the CHMTRN language for the prediction of chemical synthesis and reactions. The knowledge base contains rules for a number of endpoints including: skin sensitization, irritancy, mutagenicity, carcinogenicity and many others [7].
Utility of QSARs and expert systems [8]
A whole host of mathematical models and computational techniques are presented herein to predict metabolism and toxicity.
- The particular toxicity endpoint required.
- Whether or not a model is available and whether the training set, or knowledge base on which the model is based, is developed sufficiently for the drug in question.
- The nature of the prediction required, e.g., a quantitative or qualitative assessment of toxicity, and whether an estimate of confidence, and the level of confidence, are required. All these factors must be considered before one makes an attempt to determine whether toxicity prediction is even viable.
Disadvantages of computer-aided toxicity prediction methods [9]
The drawback with using QSARs and expert systems to predict toxicity is simple to define, yet much more complex to understand and to fix. The drawback is simple: these techniques cannot make adequate predictions for all compounds and for all endpoints, There are many reasons for this:
- Many models are poorly developed in many chemical areas. This is due to there being a paucity of available toxicity data either to build the models, or for their validation. Not only are more data required for modeling, but those data need to be of a high standard to provide reliable predictive models. An example of the gaps that are present in the training sets of the systems is that it is only OncoLogic which is able to make predictions for constituents such as fibers, polymers and inorganic containing compounds. The problem of gaps in the data will be exacerbated by the novel chemistries that are being created by combinatorial chemistry. It is unlikely that a molecular fragment rule-based approach will be able to predict reliably the toxicity of completely novel compounds [10].
- A considerable amount of expertise is required to interpret and validate the results. The basic premise of an expert system is that it presents the knowledge of an expert for use by a non-expert. It is the contention of the author that users of these systems should not, however, be non-experts. Users require an adequate level of toxicological training and expertise.
- Rule-based expert systems for predicting toxicity may be over-predictive. An example of this was the prediction of skin sensitization by the DEREK software, which predicted a number of non-sensitizers to be sensitizers as these compounds contained a structural alert. Reasons for the over-prediction include lack of knowledge concerning the effect of modulating factors on particular functional groups, and lack of permeability assessment. Whilst the latter point may be, at least partially, addressed in the StAR and HazardExpert systems, more work is required to predict membrane permeability.
- There is clearly an issue with the role of mechanisms of action in making toxicological prediction. Systems such as DEREK, HazardExpert and OncoLogic have rule bases developed specifically from a mechanistic viewpoint. Other systems such as TOPKAT and CASE are less, if at all, mechanistically based. The problems of this lack of mechanistic basis to the prediction have never, however, been adequately addressed or quantified.
- The commercial environments in which the systems are placed, effectively as competitors, does little or nothing to assist in the recognition of strengths or weaknesses of each of the systems.
- In many systems there is only limited ability to include proprietary data into the rule-base or predictive system. Some manufacturers such as CASE and TOPKAT will model proprietary data. In other systems, there are opportunities to influence the rule-base either by contributing openly to the rule base, or by the development of proprietary rule bases. Generally, though, these systems require mechanisms to allow users more freely to expand and contribute to the rule bases.
CONCLUSIONS
Computational toxicology is now widely used for lead chemical development, and are capable of providing valuable information in drug discovery process. These in silico toxicology experiments can play a major role in decreasing time to market, reducing animal experiments, assessing late stage attrition, and strategic planning of pharmaceutical and chemical development processes. Good predictive models for toxicity parameters depend crucially on selecting the right mathematical approach, the right molecular descriptors for the particular toxicity endpoint, and a sufficiently large set of experimental data relating to this endpoint for the validation of the model. In the next 10 years or so, the degree of automation of in silico modeling and data interpretation will continue to increase with the integration of medium- to high-throughput in vitro and in vivo assays to reduce the risk of late-stage attrition, and second, to optimize the screening and testing by looking at only the most promising molecules.









































































































































