Protein Structure Fingerprint Technology
- 1. Department of Medical Transformation Center, Shenzhen Institutes of Advanced Technology, China
CITATION
Yang J (2018) Protein Structure Fingerprint Technology. J Bioinform, Genomics, Proteomics 3(2): 1036
NEWS LETTER
Knowledge of Protein Structures
Knowledge of protein structures is significant for biology research and drug discovery. The protein 3D structures may be obtained by experimental measurements or computational approaches. Experimentally, the protein structures can be measured by X-ray crystallography, Nuclear Magnetic Resonance (NMR) or Transmission Electron Cryomicroscopy (Cryo TEM) etc., and so far over 140,000 of 3D structure data are available in PDB (Protein Data Bank) (https://www.rcsb.org/). Experimental measurements may accurately determine atomic coordinates of protein structures which are only some of snapshots for stable states under specific conditions. Although rich data of protein 3D structures have been accumulated by PDB, it cannot keep up with the pace of rapid increase of knowledge of protein one-dimensional sequences. Up to now, over 21,000,000 genetic codes for Prokaryotes and Eukaryota are in NCBI (National Center for Biotechnology Information) (https://www.ncbi. nlm.nih.gov/) and over 120,000,000 protein sequences in Uni Prot (Universal Protein Resource) (http://www.uniprot. org/). Overall, less than 1% of proteins have the knowledge of 3D structures. Therefore, on the other hand, the computational approaches become an important methodology to predict the protein 3D structures. Various computational methodologies for investigation of protein folding have been developed [1-4]. The comparative protein structure modeling as template-based methods to predict protein structures, which search the multiple templates with sequence homology from PDB, and then process energy optimization to predict protein 3D structure [5-7]. The ab initio or de novo approaches as template-free methods to predict protein structures, which carry out through the molecular dynamics (MD) simulation calculations under various force fields [8-14]. One large-scale project of Folding @ Home was developed in the year 2000 at Stanford University to compute the protein folding structures involving contribution from thousands of personal computer clusters world widely [15]. The CASP (platform of Critical Assessment of Techniques for protein Structure Prediction) [16] has provided a worldwide platform to promote the development of protein structure predictions since 1994.Overall, the problem of protein structure prediction is unlikely to be perfectly solved in the near future [17] because it requires vast computational resources.
Challenges in Protein Structure Research
So far, the challenges in protein structure research are in several aspects. First, most of proteins are only know one-dimensional sequences without 3D structural knowledge. Second, even if a protein 3D structure is well known, it is hard to be interpreted and analyzed due to the complexity of topological folds. Third, the nature of proteins is intrinsically disordered, and so how to present this folding flexibility. Furthermore, it is a difficult task to acquire an astronomical number of folding conformations for each protein; moreover, million numbers of proteins are waiting for solutions. Therefore, the knowledge and understanding of protein structure research is a great challenge in the field of life science in the post gene era.
Protein Structure Fingerprint Technology
A novel approach, protein structure fingerprint technology, has been developed trying to overcome these hurdles. In this approach, a folden of 5 amino acid residues was taken as the folding element, and its complete folding description were mathematically acquired. A set of 27 folding vectors was set up to fully cover various folding patterns for 5 amino acid residues, and expressed by Protein Folding Shapes Code (PFSC) as alphabetical letters [18]. Any conformation for protein three-dimensional (3D) structure can be completely presented by a PFSC string in which one PFSC letter represents for a folding vector of 5 amino acid residues, two PFSC letters next each other for coupling two folding vectors overlapping four amino acids and so on. Each set of 5 amino acid residues actually has different folding shapes and flexibilities because of molecular constrain. A database of 5 AAPFSC is created together the possible folding shapes in PFSC for various arrangements for 5 amino acids. Consequently, the comprehensive local folding variations can be obtained according the order of amino acids in sequence, and assemble in Protein Folding Variation Matrix (PFVM). The PFVM contains local folding variations along sequence. It demonstrated how the protein folding variations are determined by the order of amino acids in sequence. Also, the patterns of protein folding variations along sequence may reveal the folding flexibility and proteins intrinsic disorder. In addition, an astronomical number of protein conformations and the most possible conformations can be constructed by PFSC letters in PFVM. The protein structure fingerprint technology is briefly presented in Figure (1), and it illustrates that any protein with known 3D structure can be converted into complete conformation with PFSC description while any protein with one-dimensional sequence can acquire the comprehensive protein folding variations in PFVM (Figure 1).
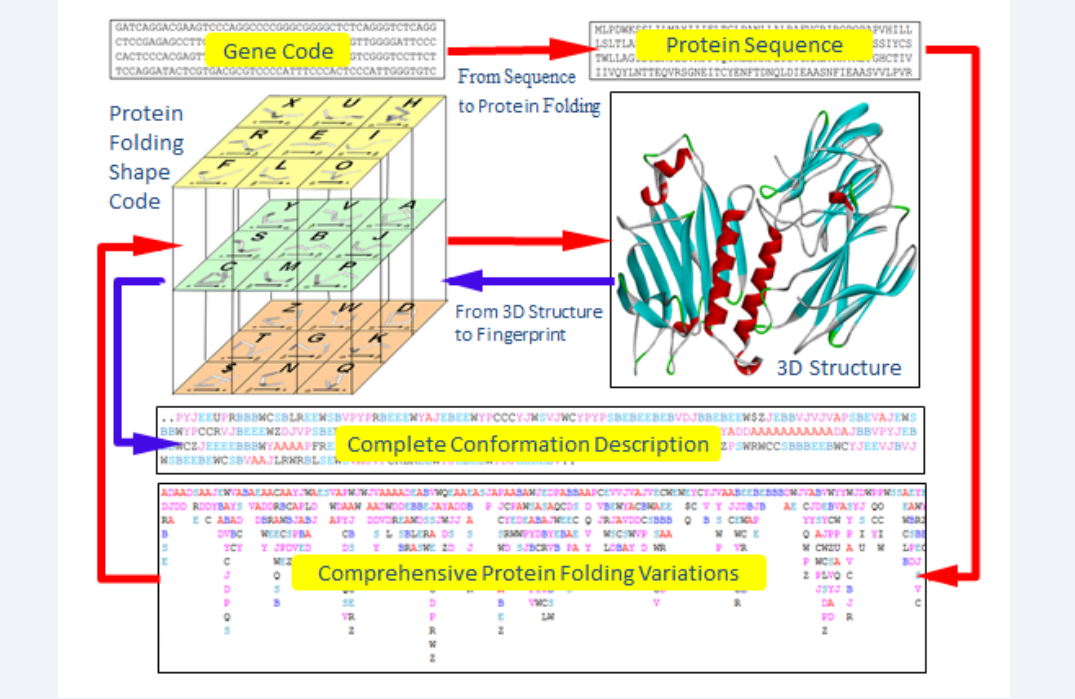
Figure 1: The protein structure fingerprint technology. 27 Protein Folding Shapes Code (PFSC) is presented in the cubic. The blue arrows indicate that the complete conformation description with using PFSC is obtained from a protein 3D structure. The red arrows indicated that the comprehensive protein folding variations in Protein Folding Variation Matrix (PFVM) is obtained from gene or protein sequence and expressed in PFSC description.
The protein structure fingerprint technology. 27 Protein Folding Shapes Code (PFSC) is presented in the cubic. The blue arrows indicate that the complete conformation description with using PFSC is obtained from a protein 3D structure. The red arrows indicated that the comprehensive protein folding variations in Protein Folding Variation Matrix (PFVM) is obtained from gene or protein sequence and expressed in PFSC description.
Web Server
A computational platform of protein structure fingerprint has been set up. If to input a protein 3D structure in PDB format, the output will present its complete conformation in PFSC string description, including secondary fragments as well as tertiary fragments. If to input an amino acid sequence, the output will provide the comprehensive protein folding variations in PFVM and further assemble the possible conformations in PFSC description. These functions can be freely accessed by web server www.micropht.com.
Potential Applications
The protein structure fingerprint technology can be applied to biology studies and drug discovery, such as gene protein mutation, protein recombination, protein differentiation analysis, protein / peptide design, antibody / antigen recognition, protein misfolding and protein stability etc. Also, it can provide abundant conformations for protein predictions.









































































































































