Virtual Screening and Molecular Docking Studies on GP1 Glycoprotein Subunit of Ebola Virus
- 1. Department of Bioinformatics, Bharathiar University,India
ABSTRACT
Ebola virus disease (EVD), also known as Ebola hemorrhagic fever is a fatal disease which spread outrageously from the month of March, 2014 on West Africa. It is very dangerous disease and no proper medicine or vaccine available till date. Experimental vaccines and treatment are under development; hence we need to find novel drug molecules having activity against Ebola virus in order to eradicate its pathogenicity. Ebola virus consists of nucleoprotein, matrix protein, non-structural proteins and structural glycoproteins. The structural glycoproteins consist of two structural subunits GP1 and GP2 which are responsible for the host cell interaction. GP1 subunit contains putative receptor regions and is responsible for host cellular attachment whereas, GP2 subunit is responsible for viral and host cell membranes fusion. The structure of GP1 subunit is taken for this study and drug like molecules which are having better binding affinity were identified through virtual screening process and molecular docking studies in order to prevent the Ebola virus in disease progression. The drug like molecule which is showing better binding affinity is suggested for further clinical trials.
KEYWORDS
• Ebola virus
• Glycoprotein
• Virtual screening
• Molecular docking
CITATION
Ruban Durairaj D, Praveen Kumar K, Lokesh T, Suganya SR, Gowdham M, Shanmughavel P (2018) Virtual Screening and Molecular Docking Studies on GP1 Glycoprotein Subunit of Ebola Virus. J Bioinform, Genomics, Proteomics 3(1): 1026.
ABBREVIATIONS
GP: Glycoprotein; EVD: Ebola Virus Disease; HTVS: High Throughput Virtual Screening
INTRODUCTION
Ebola virus is a hostile pathogen which is responsible for highly lethal hemorrhagic fever syndrome in humans which is termed as Ebola virus disease (EVD) often. The disease was first recognized in the year 1976 near the Valley of Ebola River at Zaire; hence it was named as Ebola [1]. Since then the mortality rates were ranging from 50 to 90% for the past 37 years. From 1995 to till date there are many Ebola virus disease outbreaks were identified throughout the world [2]. In recent times, the most widespread outbreak of Ebola in history was identified in the West Africa, especially in the countries of Sierra Leone, Guinea and Liberia during the year 2013-2016 [3]. The very fast disease progression of Ebola virus makes it more difficult to control this disease. Hence there is an emerging need for the identification of effective therapy of controlling and preventing the people from getting killed by this disease. There is no effective treatment or vaccines are available against the Ebola virus till date [4]. Here bioinformatics approaches and computational methods come as handy in the identification of novel drug like molecules against Ebola virus. Already few insilico findings on ebola virus matrix proteins were reported by Veljkovic., et al. [5], Karthick et al. [6] and Tamilvanan et al. [7] were provide as a supporting information of this study. HTVS (High throughput virtual screening) and molecular docking studies carried out in this study will help in identifying novel drug molecule which can be further taken for clinical trials to be actively tested as a drug [8].
Ebola virus glycoproteins
The Ebola virus consists of envelope glycoprotein (GP), Nucleoprotein (NP) and matrix proteins VP24, VP40 and nonstructural proteins including VP30 and VP35. All these proteins were encoded by genome of 19 kb long. The Ebola virus is having 2 envelope glycoproteins such as GP1 and GP2 which are playing important role in the Ebola virus disease progression [9]. The glycoproteins of Ebola virus plays a critical role in the Ebola life cycle and those GP1 and GP2 glycoproteins are the sole responsibility for the attachment, fusion and entry to the target cells. Among which GP1 subunit is highly responsible for the cellular attachment and is having well reputed receptor binding regions [10]. Hence the GP1 subunit is concentrated on this study in order to prevent the Ebola virus to getting attached with the target cell.
Virtual screening and molecular docking
High throughput virtual screening (HTVS) is a computational method to screen a high affinity molecule from the group of molecules. Literally we can say it is a method to identify one or more best molecules to be active against the target structure from the ‘n’ number of molecules [11]. A molecular docking study is a computational method to find the binding modes of a ligand with the target protein structure [12]. These 2 methods are most powerful tools in the drug designing with the aid of computers. Hence these 2 methods drastically reduce the time and cost taken in the drug designing process [13].
MATERIALS AND METHODS
3D structure retrieval of envelope glycoprotein GP1
3D structure of Ebola virus GP1 is retrieved from PDB [14] with the PDB ID of 3CSY. Resolution of the structure obtained is 3.4 Å and comprised of GP1 and GP2 glycoprotein subunits. GP1 is comprised of I, K, M, O chains whereas GP2 is comprised of J, L, N and P chains [15].
Identification of active site residues
Active site residues of the protein structure are identified by using CASTp server [16]. The residues (PHE88, ILE113, PRO116, ASP117, GLY118, SER119, GLU120, ARG136, TYR137, VAL138, HIS139, VAL141, SER142, THR144, GLY145, ARG172 and GLY173) are the active site residues of GP1 glycoprotein which are identified using CASTp server.
Retrieval of Antiviral compounds
Antiviral compounds are retrieved from PubChem database by searching the database with the keyword “antiviral” [17]. Totally 11,143 antiviral compounds were retrieved from the PubChem database and further subjected to virtual screening studies.
Preparation of Antiviral compounds
Retrieved antiviral compounds are prepared by using Ligprep [18] module of Schrodinger suite [19]. The prepared compounds are then saved and used for virtual screening process.
High throughput virtual screening (HTVS)
All the prepared ligands were further subjected to HTVS by screening the compounds in SP docking and XP docking modes available in Glide module of Schrodinger suite.
Molecular docking studies for short listed compounds
Molecular docking studies are carried out for short listed compounds using Glide module [20] of Schrodinger suite. SP docking (Standard Precision) and XP docking (Extra Precision) methods are used for identifying best molecular interaction pose of ligand with the target protein [21].
RESULTS AND DISCUSSION
HTVS (High throughput virtual screening)
Antiviral compounds retrieved from PubChem were subjected to HTVS by screening the prepared antiviral compounds by using SP and XP docking modes of Glide module in Schrodinger suite. The compounds were shortlisted by applying Lipinski rule of 5. The compounds passing all these criteria were taken for further molecular docking simulation studies. Totally 1,232 compounds were passed all these criteria and further taken for the molecular docking studies.
SP (Standard precision) docking
Shortlisted compounds satisfying the Lipinski rule of 5 were further taken for SP docking. The top 10 compounds from the SP docking and its scores are displayed in the Table 1.
XP (Extra precision) docking
Compounds passed from SP docking are further taken for XP docking in order to remove false positives. The top 10 compounds from the XP docking and its scores are displayed in the Table 2.
Molecular docking interaction
The top ranked compound which passed in both SP and XP docking mode is further taken for Molecular docking interaction study. Glide module of Schrodinger suite is used for the molecular docking studies. The molecular docking interactions between the compound and GP1 are described in the Table 3. 3D and 2D representation of molecular interaction between the compound and GP1 are depicted in the Figures 1 and 2 respectively.
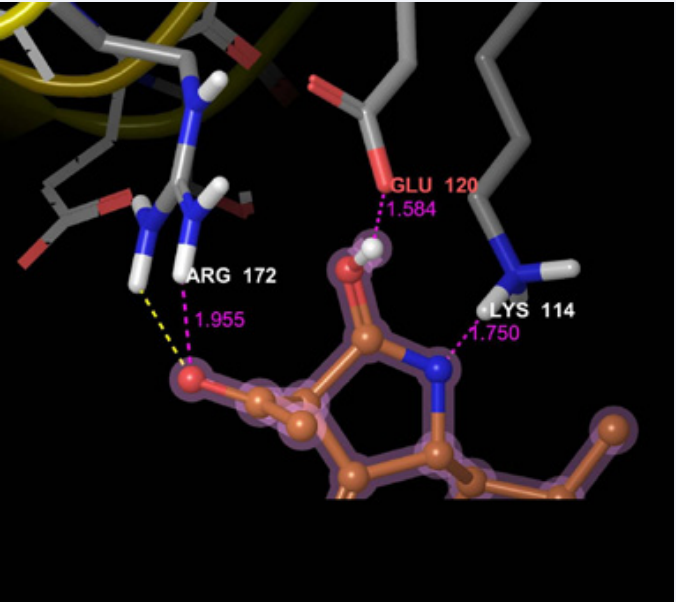
Figure 1: 3D representation of molecular docking interactions of compound 54737266 with Ebola virus envelope Glycoprotein GP1.
Figure 2: 2 dimensional representation of molecular docking interaction of compound 54737266 with Ebola virus envelope Glycoprotein GP1.
CONCLUSION
From this study by combining the virtual screening, molecular docking and molecular dynamics simulation studies, the Pubchem compound 54737266 is showing better binding activity with the Ebola virus envelope glycoprotein GP1. And also the PubChem compound is satisfying Lipinski’s rule of 5 and ADMET properties, to be a drug. Hence it can be used as a novel ligand in further drug discovery process for treating the Ebola virus disease.
Table 1: SP docking results of top 10 antiviral compounds of PubChem showing activity against the GP1 subunit.
| Sl. No | Compound ID | Docking score | Glide energy |
| 1 | 54737266 | -5.20391 | -28.410878 |
| 2 | 5351180 | -4.690352 | -28.726926 |
| 3 | 102198 | -4.555893 | -28.435244 |
| 4 | 96368 | -4.37506 | -27.268888 |
| 5 | 45038823 | -4.326942 | -21.515891 |
| 6 | 54678599 | -4.297013 | -15.936589 |
| 7 | 37542 | -4.272528 | -27.389997 |
| 8 | 50599 | -4.223172 | -23.389505 |
| 9 | 3043 | -4.220418 | -23.386317 |
| 10 | 64987 | -4.162647 | -30.688543 |
| Abbreviations: SP: Standard Precision; GP1: Glycoprotein1 | |||
Table 2: XP docking results of top 10 antiviral compounds of PubChem showing activity against the GP1 subunit.
| Sl. No | Compound ID | Glide energy | XP GScore |
| 1 | 54737266 | -14.872 | -5.28141 |
| 2 | 45038823 | -19.8117 | -4.27847 |
| 3 | 34768 | -23.232 | -4.34689 |
| 4 | 5064 | -28.538 | -3.94815 |
| 5 | 336497 | -24.7215 | -3.86519 |
| 6 | 54683011 | -15.582 | -3.88903 |
| 7 | 54723289 | -14.5674 | -3.87609 |
| 8 | 439268 | -17.4376 | -3.75795 |
| 9 | 102198 | -28.0309 | -3.66702 |
| 10 | 5351180 | -28.5095 | -3.59863 |
| Abbreviations: XP: Xtra Precision; GP1: Glycoprotein1 | |||
Table 3: Molecular docking interaction of the PubChem compound 54737266 against the GP1 subunit of Ebola virus.
| Compound ID | Docking score | Glide energy | Interacting residues | Bond length Å |
| 54737266 | -5.256 | -14.872 | LYS 114 | 1.750 |
| GLU 120 | 1.584 | |||
| ARG 172 | 1.955, 2.365 |
ACKNOWLEDGEMENTS
We acknowledge DBT-Centre for Bioinformatics, Bharathiar University, Coimbatore, Tamilnadu, India for providing all the computational facilities to carry out this study successfully.









































































































































