Computational Analysis of Single Nucleotide Polymorphism (Snps) In Human MYC Gene
- 1. Department of Bioinformatics, Africa city of technology, Sudan
- 2. Department of Bioinformatics, Ibn Sena University, Sudan
- 3. Department of Bioinformatics, Omdurman Ahlia University, Sudan
- 4. Department of Bioinformatics, Omdurman Islamic University, Sudan
- 5. Department of Bioinformatics, Alribat hospital University, Sudan
ABSTRACT
Background: The proto-oncogene c-MYC encodes a transcription factor that regulates cell proliferation, growth, apoptosis microRNAs expression. Dysregulated expression or function of c-Myc is one of the most common abnormalities in human malignancy. The c-myc gene comprises three exons. Exon 1 contains two promoters and is non coding. Exons 2 and 3 encode the Myc protein with translation initiation at nucleotide 16 of exon. In this paper we focused on predicting the effects that can be imposed by single nucleotide polymorphisms that have been reported in MYC gene using Insilico approaches.
Material and Methods: MYC gene was investigated in NCBI database (http://www.ncbi.nlm.nih.gov/) and SNPs were analyzed by computational softwares. SNPs in the coding region (exonal SNPs) that are non-synonymous (nsSNP) were analyzed by (sift, polyphen, Imutant and PHD-snp) softwares, and SNPs at un-translated region at 3’ends (3’UTR) were analyzed to predict the effect on miRNA binding on these regions that may greatly associated with tumor progression [25]. The SNPs at un-traslated region at 5’ ends (5UTR) were analyzed too by SNPs Function prediction software
Result: We analyzed 5954 SNPs from NCBI ,647 of them found in Homo sapiens, 156 SNPs in coding non synonymous regions (missense), 101 synonymous, 42 3UTR and 47 5UTR. Only SNPs are present on coding region, 3UTR and 5UTR selected to analysis.
Conclusion: Four SNPs had high score with PSIC SD range (1-099) and TOLERANCE INDEX equal (0 - 0.009); rs200431478, rs114570780, rs150308400, rs137906262. There were predicted to change the protein stability but only rs150308400 was predicted to be disease related. in 3UTR there were only 11 functional SNPs predicted, rs185650723 and rs4645970 contain D allele which is derived allele that disrupts a conserved miRNA sit while rs35524866 SNP contain (C) allele which is can create a new microRNA site.
KEYWORDS
• Proto-oncogene
• Malignancy
• Insilico
• Synonymous
CITATION
Fadlalla Elshaikh AAE, Elmahdi Ahmed MT, Daf Alla TIM, Mogammed Elbasheer AS, Ahmed AA, et al. (2016) Computational Analysis of Single Nucleotide Polymorphism (Snps) In Human MYC Gene. J Bioinform, Genomics, Proteomics 1(3): 1011.
INTRODUCTION
Lymphomas are a group of diseases caused by malignant lymphocytes that accumulate in lymph nodes and cause the characteristic clinical features of lymphadenopathy. Occasionally, they may spill over into blood or infiltrate organs outside the lymphoid tissue [1]. Burkitt lymphoma, a subdivision of lymphoma, is particularly prevalent in young children in tropical Africa, accounting for 30%–50% of all childhood cancers in equatorial Africa, most frequently affect extranodal sites including the jaws, the abdomen, and endocrine organs [2]. The disease is heterogeneous and harbouring many genetic abnormalities including disruption of C-Myc gene.
The proto-oncogene c-MYC encodes a transcription factor that regulates cell proliferation, growth, apoptosis [3] microRNAs expression [4,5]. Also it can facilitate mRNA cap methylation and translation [6] and stimulates transcription of rRNA genes [7]. Dysregulated expression or function of c-Myc is one of the most common abnormalities in human malignancy [8]. The c-myc gene comprises three exons. Exon 1 contains two promoters and is non coding. Exons 2 and 3 encode the Myc protein with translation initiation at nucleotide 16 of exon 2 [9].
A defining feature of Burkitt lymphoma is activation of the MYC gene at 8q24 through translocation with one of three immunoglobulin loci, which introduces a transcriptional enhancer element. In 80% of cases, this involves the immunoglobulin heavy chain locus at 14q32, with the breakpoint in the class switch region. In 15%, the gene encoding the kappa light chain at 2p11 is involved; while in 5% the lambda light chain gene at 22q11 is translocated resulting in overproduction of MYC protein [10,11].
Given its pivotal above mentioned roles many studies have focused on studying myc extensively especially the translocation. Myc transcriptional activity is regulated by phosphorylation at Ser-62 followed by Thr-58, and subsequent proteasomal degradation after performing its function [12-16]. Mutations of Myc residues Thr-58 and Ser-62, prevalently found in Burkitt lymphoma, are associated with stabilized mutant protein. In this paper we focused on predicting the effects that can be imposed by single nucleotide polymorphisms that have been reported in myc gene using Insilico approaches to shed a light on effect of these polymorphisms as the MYC protein levels are critically regulated, and even relatively small increases can destabilize cell growth control.
MATERIALS AND METHODS
The critical step in this work was to select SNPs for analysis by computational softwares; the selection was prioritizing SNPs in the coding region (exonal SNPs) that are non-synonymous (nsSNP) and SNPs at un-translated region at 3’ends (3’UTR) to predict the effect on miRNA binding on these regions that may greatly associated with tumor progression [13]. The SNPs at un-traslated region at 5’ ends (5UTR) were analyzed too by SNPs Function prediction software. The SNPs and the related ensembles protein (ESNP) were obtained from the SNPs database (dbSNPs) for computational analysis from http://www.ncbi.nlm. nih.gov/snp/ and Uniprot database.
GeneMANIA
GeneMANIA (http://www.genemania.org) is a web interface that helps predicting the function of genes and gene sets. GeneMANIA finds other genes that are related to a set of input genes, using a very large set of functional association data. Association data include protein and genetic interactions, pathways, co-expression, co-localization and protein domain similarity. GeneMANIA can be used to find new members of a pathway or complex, find additional genes you may have missed in your screen or find new genes with a specific function, such as protein kinases. Your question is defined by the set of genes you input [17].
Sorting intolerant from tolerant (SIFT)
SIFT (http://siftdna.org/www/SIFT_dbSNP.html) predicts the tolerated and deleterious SNPs and identifies the impact of amino acid substitution on protein function and phenotype alterations, so that users can prioritize substitutions for further study. The main underlying principle of this program is that it generates alignments with a large number of homologous sequences, and assigns scores to each residue ranging from zero to one. The threshold intolerance score for SNPs is 0.05 or less [18,19].
PolyPhen
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) is an online bioinformatics program to automatically predict the consequence of an amino acid change on the structure and function of a protein. This prediction is based on a number of features comprising the sequence, phylogenetic and structural information characterizing the substitution. Basically, this program searches for 3D protein structures, multiple alignments of homologous sequences and amino acid contact information in several protein structure databases, then calculates position-specific independent count scores (PSIC) for each of the two variants, and then computes the PSIC scores difference between two variants. The higher a PSIC score difference, the higher the functional impact a particular amino acid substitution is likely to have. Prediction outcomes could be classified as benign, possibly damaging or probably damaging, according to the posterior probability intervals (0, 0.2), (0.2, 0.85) and (0.85, 1), respectively. nsSNPs that predicted to be intolerant by Sift has been submitted to Polyphen as protein sequence in FASTA format that obtained from UniproktB/Expasy after submitting the relevant ensemble protein (ESNP) there, then we entered position of mutation, native amino acid and the new substituent for both structural and functional predictions [20].
I-Mutant
I-Mutant version 3.0 (http://gpcr2.biocomp.unibo.it/cgi/ predictors/I-Mutant3.0/I-Mutant3.0.cgi) was used to predict the protein stability changes upon single-site mutations. I-Mutant basically can evaluate the stability change of a single site mutation starting from the protein structure or from the protein sequences [21].
Predictor of human deleterious single nucleotide polymorphisms (PHD-SNP)
PhD- SNP is a web-based tool available at (http://snps. biofold.org/phd-snp/phd-snp.html).It predicts whether the new phenotype derived from a nsSNP is a disease related or not (neutral). Protein sequence from uniprot is submitted to the program after providing position and the new amino acid residue [22].
Project HOPE
Project Have Our Protein Explained (HOPE; http://www. cmbi.ru.nl/hope/home) is an automatic mutant analysis server to study the insight structural features of native protein and the variant models. HOPE provides the 3D structural visualization of mutated proteins, and gives the results by using UniProt and DAS prediction servers. Input method of Project HOPE carries the protein sequence and selection of Mutant variants. HOPE server predicts the output in the form of structural variation between mutant and wild type residues [23]. We submitted a sequence and mutation only for those that predicted to be damaging by both SIFT and Polyphen (Double Positive) servers.
Raptorx
It is Web-based method for protein secondary structure prediction, (http://raptorx.uchicago.edu/). It based on tertiary structure modeling, alignment quality assessment and sophisticated probabilistic alignment sampling. raptorX delivers high-quality structural models for many targets and it takes 35 min to finish processing a sequence of 200 amino acids [24]. C-MYC Protein sequences of the most deletrious nsSNP were presented to raptorx server to get the model sequence as PDB file. After that Chimera program had been to visualize the PDB file.
Chimera
Chimera (http://www.cgl.ucsf.edu/chimera) is a high-quality extensible program for interactive conception and analysis of molecular assemblies and related data [25]. This software produced by University of California, San Francisco (UCSF). Chimera (version 1.6.2) was used to generate the mutated 3D model models of each C-Myc protein [26].
PolymiRTS
(http://compbio.uthsc.edu/miRSNP/) is the database server designed specifically for the analysis of the 3’UTR region; at this stage we used this server to determine SNPs that may alter miRNA target sites. All SNPs located within the 3′-UTRs of database were selected separately and submitted to the program. Then we checked if the SNP variants could alter putative miRNA target sites focusing on mutations that alter sequence complementarity to miRNA seed regions [27].
SNP Function Prediction
(https://snpinfo.niehs.nih.gov/snpinfo/snpfunc.htm) It Is software designed to be a clearing house for all public domain SNP functional annotation data, as well as in-house functional annotations derived from different data sources. It currently contains SNP functional annotations in six major categories including genomic elements, transcription regulation, protein function, pathway, disease and population genetics [28].
RESULTS AND DISCUSSION
MYC gene has a vital role in human body and it is co-expressed with 14 genes listed in Table (1) and shared domain with only one gene (MAX) gene (Figure 1).
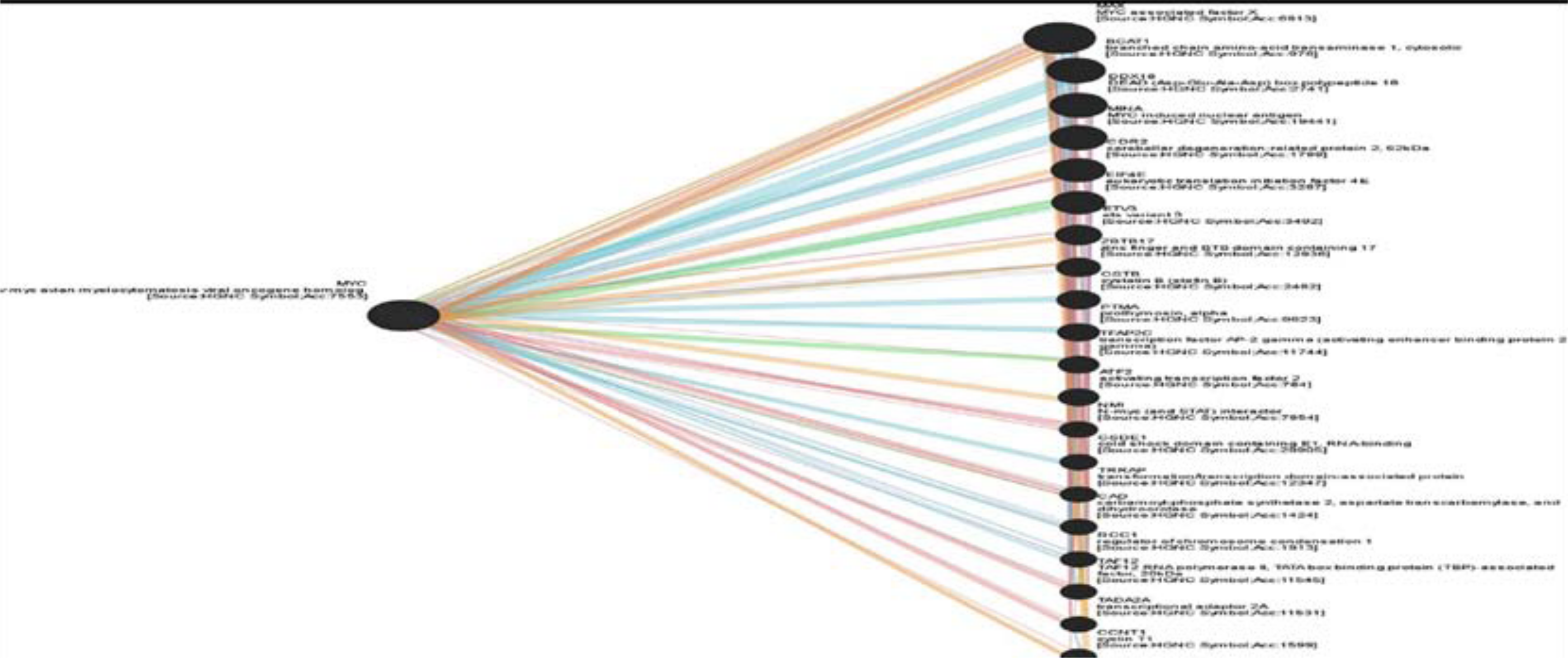
Figure 1: Show genes co- expression with C-MYC gene.
MYC gene was investigated in NCBI database (http://www.ncbi.nlm.nih.gov/). It contains a total of 5954 SNPs and 647 of which on Homo sapiens, 156 coding non synonymous regions (missense), 101 synonymous, 42 3UTR and 47 5UTR. Only SNPs are present on coding region, 3UTR and 5UTR selected to analysis. Non synonymous SNPs were analyzed by SIFT software, out of 29 SNPs only 15 SNPs were predicted to be deleterious. These deleterious SNPs were analyzed using PolyPhen software to predict the damaging SNPs, we found that 10 SNPs were predicted to be deleterious in both softwares. Four SNPs had high score with PSIC SD range (1- 099) and TOLERANCE INDEX equal (0 - 0.009); rs200431478, rs114570780, rs150308400, rs137906262 Table (3), Figure (2).
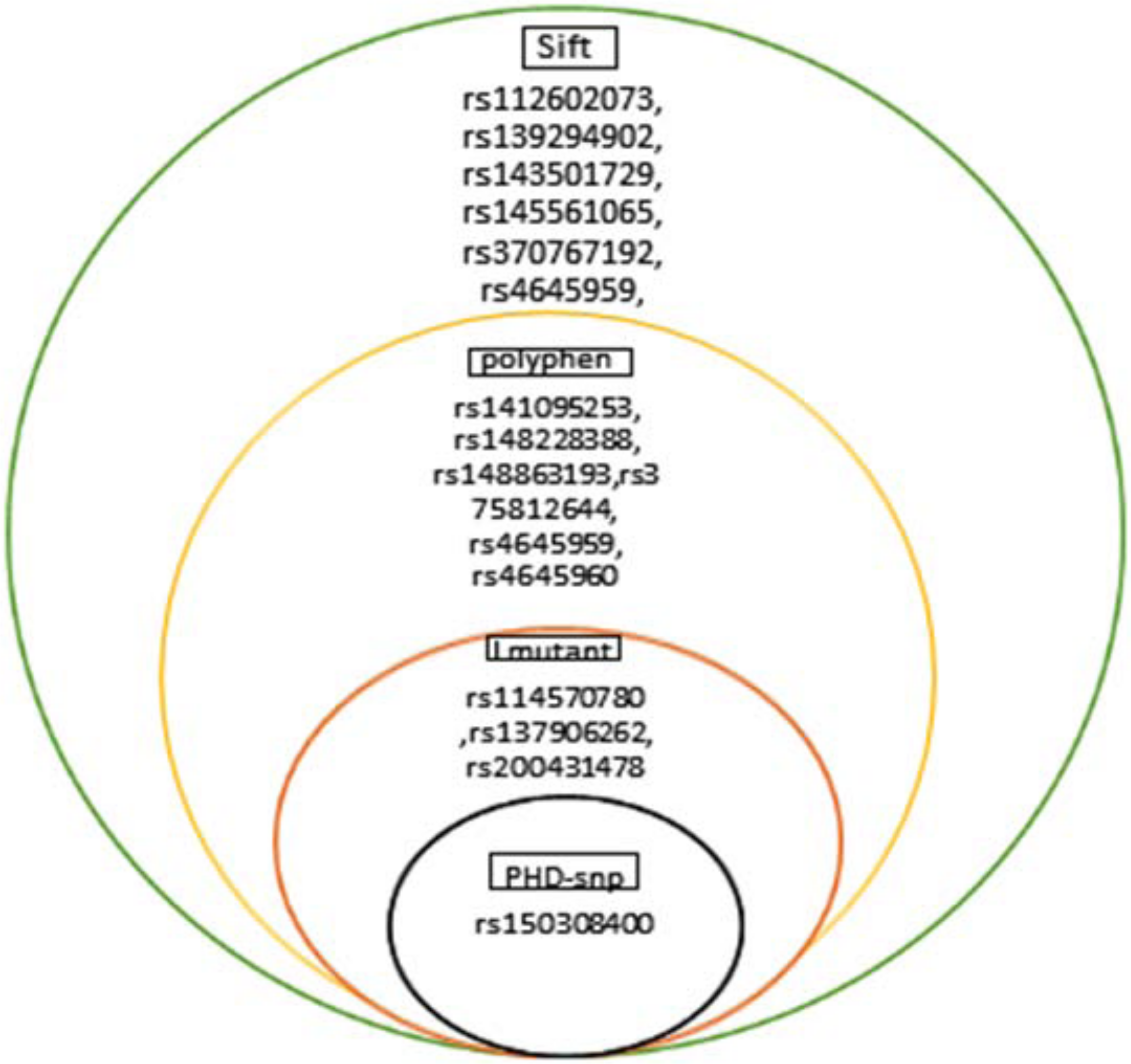
Figure 2: Show SNPs of human c-Myc1 protein predicted by SIFT, PolyPhen, Imutant and PHD-snp.
The same result was predicted by Mamoona Noreen et. al in 2015 [29].The rs200431478 result in substitution of a serine into a bigger and more hydrophobic phenylalanine at position (362 and 361) causing bumps and loss of hydrogen bonds, change of secondary structure, slight conformation destabilized and disturb correct folding and phosphorylation modification sit according to project hope software. The rs150308400 caused conversion of amino acid cysteine with a bigger and less hydrophobic tyrosine at position (148, 133 and 147) leading to bumps and loss of hydrophobic interactions and it was predicted to be disease related by PHD-snp software. These two SNPs were predicted to increase effective stability of protein using I mutant software Figure (3).
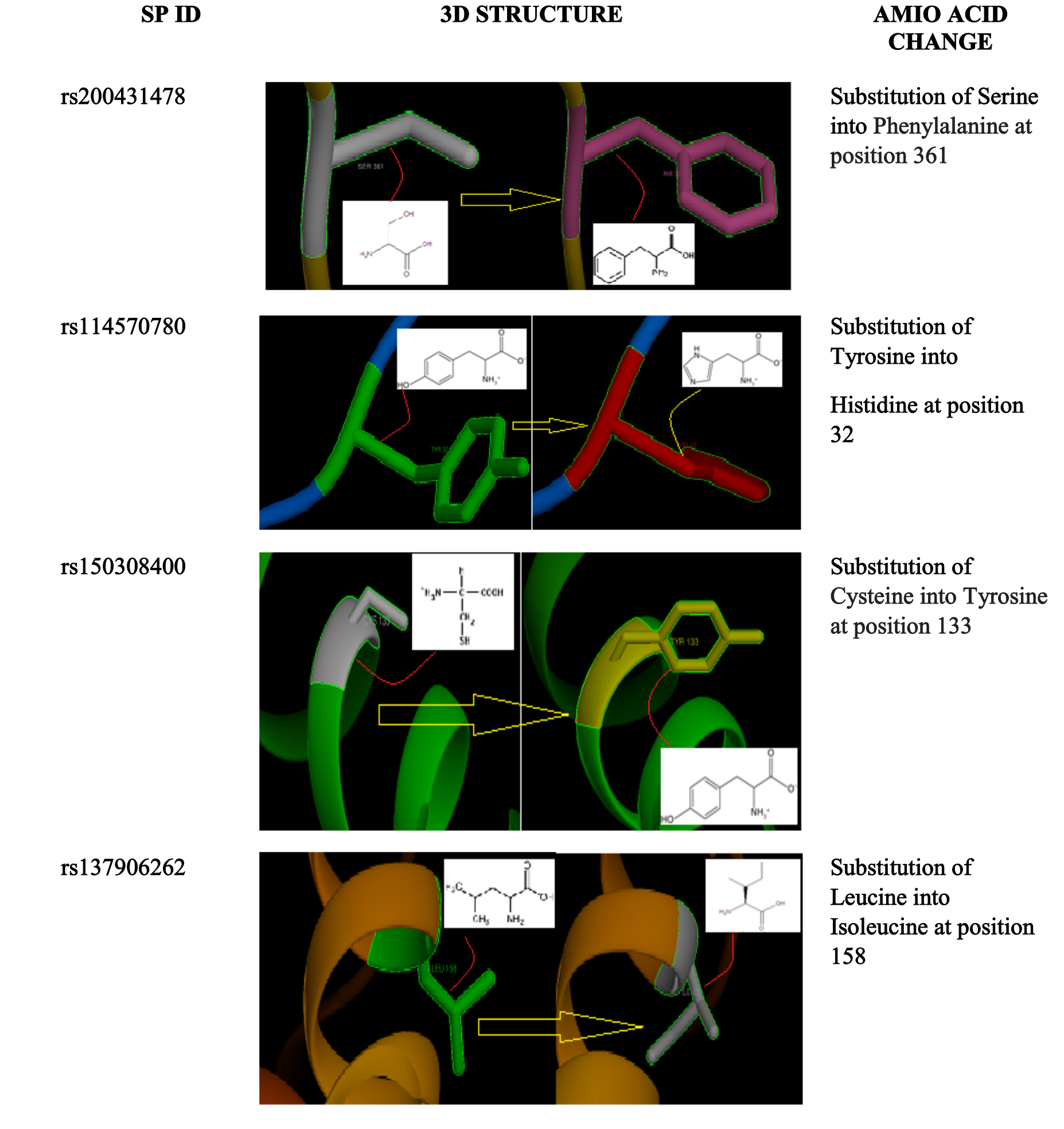
Figure 3: 3D model by Chimera for MYC protein.
The rs114570780 result in replacement of a tyrosine with a histidine at position (47, 46 and 32) which lead to loss of interactions, loss of hydrophobic interactions and disturbance the site of modification owing to histidine is smaller, less hydrophobic and not provide phosphorylation site according to Project hope software. This SNP was predicted to decrease effective stability of protein by I mutant software (Figure 3). The was no difference between the two amino acid in the fourth SNP rs137906262, leucine into isoleucine at position 158, but the mutant residue might disturb Sequence-Specific DNA Binding Transcription Factor Activity according to project hope software. This SNP also was predicted to decrease effective stability of protein by I mutant software (Figure 3). Functional SNPs in 3 untranslated region in MYC gene was analyzed using PollymiRTS software. Among 42 SNPs in 3UTR there were only 11 functional SNPs predicted. Rs 185650723 SNP contain (D) allele have (4) miRNA Site and rs4645970 SNP contain (D) allele have (4) miRNA Site which they are derived allele that disrupts a conserved miRNA sit. Rs35524866 SNP contain (C) allele have 5 miRNA Site as Target binding site can create a new microRNA site Table (4).
Table 1: shows the genes co-expressed and share a domain with C-MYC.
| Gene Symbol | Description | CO-EX-PRESSION | Shared domain |
| MAX | MYC associated factor X | NO | Yes |
| BCAT1 | branched chain amino-acid transaminase 1, cytosolic | NO | NO |
| DDX18 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 18 | YES | NO |
| MINA | MYC induced nuclear antigen | YES | NO |
| CDR2 | cerebellar degeneration-related protein 2, | YES | NO |
| EIF4E | eukaryotic translation initiation factor 4E | YES | NO |
| ETV3 | ets variant 3 | NO | NO |
| ZBTB17 | zinc finger and BTB domain containing 17 | YES | NO |
| CSTB | cystatin B (stefin B) | YES | NO |
| PTMA | prothymosin, alpha | NO | NO |
| TFAP2C | transcription factor AP-2 gamma (activating enhancer binding protein 2 gamma) | NO | NO |
| ATF2 | activating transcription factor 2 | YES | NO |
| NMI | N-myc (and STAT) interactor | YES | NO |
| CSDE1 | cold shock domain containing E1, RNA-binding | YES | NO |
| TRRAP | transformation/transcription domain-associated protein | YES | NO |
| CAD | carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase | YES | NO |
| RCC1 | regulator of chromosome condensation 1 | YES | NO |
| TAF12 | TAF12 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 20kDa | YES | NO |
| TADA2A | transcriptional adaptor 2A | YES | NO |
| CCNT1 | cyclin T1 | NO | NO |
Table 2: shows the C-MYC functions and its appearance in network and genome.
| Feature | FDR | Genes in network | Genes in genome |
| internal peptidyl-lysine acetylation | 1.98E-02 | 4 | 102 |
| SAGA-type complex | 1.98E-02 | 3 | 27 |
| histone acetylation | 1.98E-02 | 4 | 101 |
| internal protein amino acid acetylation | 1.98E-02 | 4 | 108 |
| peptidyl-lysine acetylation | 1.98E-02 | 4 | 106 |
| protein acetylation | 2.60E-02 | 4 | 121 |
| histone acetyl transferase activity | 2.94E-02 | 3 | 40 |
| peptidyl-lysine modification | 3.28E-02 | 4 | 138 |
| protein acylation | 3.64E-02 | 4 | 146 |
| N-acetyltransferase activity | 4.84E-02 | 3 | 53 |
| N-acyltransferase activity | 5.48E-02 | 3 | 57 |
| acetyltransferase activity | 6.47E-02 | 3 | 62 |
| histone acetyltransferase complex | 8.23E-02 | 3 | 69 |
| STAGA complex | 9.28E-02 | 2 | 12 |
| protein acetyltransferase complex | 9.28E-02 | 3 | 77 |
| acetyltransferase complex | 9.28E-02 | 3 | 77 |
| FDR: False discovery rate is greater than or equal to the probability that this is a false positive | |||
Table 3: shows of nonsynonymous SNPs predicted with SIFT, Polyphen, I-Mutant and PHD-snp programs, chosen SNPs with PSIC SD range (1-099) and TOLERANCE INDEX equal (0.009).
| I mutant | PHD-SNP | ||||||||||||
| SNP | PROTEIN ID | REF ALLELE | ALT ALLELE | AMINO ACID CHANGE | SIFT PREDICTION | SIFT SCORE | polyphen prediction | polyphen score | SVM2 Prediction Effect | DDG Value Prediction | RI | effect | RI |
| rs200431478 | ENSP00000367207 | C | T | S362F | DELETERIOUS | 0.003 | probably damaging | 0.998 | Increase | -0.09 | 5 | Neutral | 5 |
| rs200431478 | ENSP00000430235 | C | T | S361F | DELETERIOUS | 0.003 | probably damaging | 0.998 | Increase | -0.09 | 5 | Neutral | 5 |
| rs114570780 | ENSP00000259523 | T | C | Y32H | DELETERIOUS | 0 | probably damaging | 0.999 | Decrease | -0.63 | 1 | Neutral | 6 |
| rs114570780 | ENSP00000367207 | T | C | Y47H | DELETERIOUS | 0 | probably damaging | 0.996 | Decrease | -0.63 | 1 | Neutral | 6 |
| rs114570780 | ENSP00000430235 | T | C | Y46H | DELETERIOUS | 0 | probably damaging | 1 | Decrease | -0.63 | 1 | Neutral | 6 |
| rs150308400 | ENSP00000259523 | G | A | C133Y | DELETERIOUS | 0 | probably damaging | 0.996 | Increase | -0.23 | 1 | Disease | 9 |
| rs150308400 | ENSP00000367207 | G | A | C148Y | DELETERIOUS | 0 | probably damaging | 0.991 | Increase | -0.23 | 1 | Disease | 9 |
| rs150308400 | ENSP00000429441 | G | A | C147Y | DELETERIOUS | 0 | probably damaging | 0.999 | Increase | -0.23 | 1 | Disease | 9 |
| rs137906262 | ENSP00000429441 | C | A | L158I | DELETERIOUS | 0.009 | possibly damaging | 0.933 | Decrease | -1 | 6 | Neutral | 2 |
Table 4: shows the SNPs predicted by Polymirt to induce disruption or formation of mirRNA binding site:
| dbSNP ID | Variant | Wobble | Ancestral | Allele | miR ID | Conservation | miRSite | Function | context+ | |
| type | base pair | Allele | Class | score change | ||||||
| 1.29E+08 | rs200447778 | SNP | Y | A | ||||||
| G | hsa-miR-1238-3p | 7 | aaaagtGAGGAAA | C | No Change | |||||
| hsa-miR-670-3p | 6 | aaaagtGAGGAAA | C | No Change | ||||||
| 1.29E+08 | rs181048497 | SNP | N | C | ||||||
| G | hsa-miR-1178-3p | 1 | aatgtcGTGAGCA | C | -0.267 | |||||
| 1.29E+08 | rs35524866 | SNP | Y | G | ||||||
| A | hsa-miR-219a-5p | 1 | tcctgaACAATCA | C | -0.096 | |||||
| hsa-miR-4445-5p | 1 | tcctgAACAATCA | C | -0.228 | ||||||
| hsa-miR-4782-3p | 1 | tcctgaACAATCA | C | -0.096 | ||||||
| hsa-miR-508-3p | 1 | tcctgaACAATCA | C | -0.097 | ||||||
| hsa-miR-6766-3p | 1 | tcctgaACAATCA | C | -0.096 | ||||||
| 1.29E+08 | rs200570465 | SNP | N | A | ||||||
| C | C hsa-miR-24-3p | 1 | CTGAGCCAtcacc | C | -0.402 | |||||
| hsa-miR-4284 | 1 | cTGAGCCAtcacc | C | -0.163 | ||||||
| 1.29E+08 | rs2070583 | SNP | Y | A | A | hsa-miR-6800-5p | 2 | gcaaTCACCTAtg | D | -0.298 |
| G | hsa-miR-8053 | 1 | gcAATCGCCtatg | C | -0.298 | |||||
| 1.29E+08 | rs149534345 | SNP | Y | A | ||||||
| G | hsa-miR-4677-3p | 15 | CTCACAGccttgg | C | -0.135 | |||||
| hsa-miR-7974 | 19 | ctCACAGCCttgg | C | -0.236 | ||||||
| 1.29E+08 | rs14607 | SNP | N | T | T | hsa-miR-4432 | 3 | GAGTCTTgagact | D | -0.177 |
| hsa-miR-513c-5p | 2 | gagtCTTGAGAct | D | -0.13 | ||||||
| hsa-miR-514b-5p | 2 | gagtCTTGAGAct | D | -0.148 | ||||||
| C | hsa-miR-516a-5p | 2 | gagtCTCGAGAct | C | -0.297 | |||||
| 1.29E+08 | rs185650723 | SNP | N | C | C | hsa-miR-135a-5p | 13 | atttAGCCATAat | D | -0.17 |
| hsa-miR-135b-5p | 13 | atttAGCCATAat | D | -0.17 | ||||||
| hsa-miR-8074 | 11 | atttaGCCATAAt | D | -0.197 | ||||||
| hsa-miR-889-5p | 13 | atttAGCCATAat | D | -0.164 | ||||||
| T | hsa-miR-6831-3p | 12 | atTTAGTCAtaat | C | -0.131 | |||||
| 1.29E+08 | rs4645970 | SNP | Y | A | A | hsa-miR-135a-5p | 13 | ttAGCCATAatgt | D | -0.17 |
| hsa-miR-135b-5p | 13 | ttAGCCATAatgt | D | -0.17 | ||||||
| hsa-miR-8074 | 11 | ttaGCCATAAtgt | D | -0.197 | ||||||
| hsa-miR-889-5p | 13 | ttAGCCATAatgt | D | -0.164 | ||||||
| G | hsa-miR-323a-3p | 14 | ttagccGTAATGT | C | -0.151 | |||||
| 1.29E+08 | rs143895359 | SNP | N | A | ||||||
| 1.29E+08 | rs190322311 | SNP | Y | A | ||||||
| G | hsa-miR-1284 | 2 | cctagTGTATAGt | C | -0.152 | |||||
| hsa-miR-4704-5p | 1 | cCTAGTGTAtagt | C | -0.386 | ||||||
| hsa-miR-4789-5p | 1 | cctaGTGTATAgt | C | -0.147 |
Table 5: shows the SNPs predicted by SNPs Function prediction in 5UTR.
| SNP | Allele | Position | Prediction Strand | Forward Sequence | Matrix | Score | Method |
| rs4645946 | G | 2 | + | CGAGAAG | SRp40 | 2.86 | ESEfind |
| rs4645946 | A | 4 | + | CTCAAGA | SF2ASF2 | 3.07 | ESEfind |
| rs4645946 | A | 4 | + | CTCAAGA | SF2ASF1 | 2.95 | ESEfind |
| rs4645946 | A | 5 | + | CCTCAAG | SRp40 | 4.35 | ESEfind |
| rs4645946 | G | 1 | + | GAGAAG | NA | NA | RESCUE-ESE |
| rs4645946 | A | 1 | + | AAGAAG | NA | NA | RESCUE-ESE |
| rs4645946 | A | 2 | + | CAAGAA | NA | NA | RESCUE-ESE |
| rs4645946 | A | 3 | + | TCAAGA | NA | NA | RESCUE-ESE |
REFERENCES
1. Hoffbrand AV, Pettit JE, Moss PAH. Essential haematology. 6th edition, Blackwell science Inc.2011.
3. Levens D. Disentangling the MYC web. Proc Natl Acad Sci U S A. 2002; 99: 5757-5759.
5. Dang CV. MYC on the path to cancer. Cell. 2012; 149: 22-35.
10. Archibald S. Perkins, Jonathan W. Friedberg. Burkitt Lymphoma in Adults hematology. 2008; 341-348.









































































































































{kind=link}