Concomitant Acquired Hemophilia A and Acquired von Willebrand Syndrome from Distinctive Autoantibodies: Case Report
- 1. Western University, Department of Medicine, USA
- 2. Queens University, Department of Medicine, USA
- 3. University of Montreal, Department of Hematology, USA
Abstract
Acquired Hemophilia A (AHA) and Acquired von Willebrand Syndrome (AVWS) are rare bleeding disorders that do not often present concurrently. Here, we report a coexisting AHA and AVWS case due to underlying autoantibodies to Factor VIII (FVIII) and von Willebrand Factor (VWF). A patient with GI bleed and prolonged aPTT was diagnosed with AHA and AVWS. The patient was started on immunosuppression with prednisone, cyclophosphamide and intravenous immunoglobulin, alongside recombinant porcine FVIII replacement, susoctocog alfa (Obizur®). AVWS reduced the half-life of susoctocog alfa, requiring more frequent dosing and laboratory monitoring until AVWS resolved. The patient relapsed after three weeks, required Rituximab, and has been maintained in remission since. Given the potential therapeutic implications, VWF testing should be considered as part of the diagnostic workup for AHA.
Keywords
• Acquired Hemophilia A
• Acquired Von Willebrand Syndrome
• Autoantibody
• Case Report
Citation
Yu R, Bowman M, Bonnefoy A, James P, Phua CW (2025) Concomitant Acquired Hemophilia A and Acquired von Willebrand Syndrome from Distinctive Autoantibodies: Case Report. JSM Clin Case Rep 13(2): 1259.
ABBREVIATIONS
AHA: Acquired Hemophilia A; AVWS: Acquired Von Willebrand Syndrome; BU: Bethesda Unit; ELISA: EnzymeLinked Immunosorbent Assay; GPIb: Glycoprotein Ib; IVIG: Intravenous Immunoglobulins; PRBC: Packed Red Blood Cells; APTT: Activated Partial Thromboplastin Time; STEMI: ST-Elevation Myocardial Infarction; VWFpp: VWF Propeptide Antigen
INTRODUCTION
Acquired Hemophilia A (AHA) is a rare disorder with a reported incidence of 1.0–1.5 case per million [1]. It is due to autoantibodies that target factor VIII (FVIII), causing FVIII deficiency and, consequently, bleeding. Etiologies of AHA are varied; fifty percent are idiopathic. Other associated conditions include monoclonal gammopathy, lymphoproliferative disorder, autoimmune conditions (e.g. systemic lupus), or medications [2]. Acquired von Willebrand Syndrome (AVWS) is another rare bleeding disorder characterized by deficiency and/ or dysfunction of von Willebrand Factor (VWF). Etiologies include autoantibodies that directly inhibit the activity of VWF or increase clearance from circulation [3], shearstress proteolysis (E.g. Aortic stenosis, cardiac devices) [4], and adsorption of VWF onto cells that express the VWF receptor glycoprotein Ib (e.g. Wilm’s tumours) [5]. A single-center Mayo Clinic study estimated a prevalence of 0.04% (1 per 2500 patients) [6]. We present a case of concomitant AHA and AVWS, driven by the production of two autoantibodies with distinct epitope specificities and their significant clinical implications.
CASE PRESENTATION
A 73-year-old Caucasian female with a history of stroke, St-Elevation Myocardial Infarction (STEMI) on aspirin, and collagenous colitis presented to the hospital in 2023 after one week of melena stools with chest pain from demand ischemia. Her hemoglobin was low at 49 g/L, necessitating transfusions of 3 units of Packed Red Blood Cells (PRBCs). Aside from pallor, her physical exam was largely unremarkable, with no signs of gingival bleeding, petechiae, or hemarthrosis. Further bloodwork with reference ranges is summarized in Table 1.
Table 1: Summary of Baseline and Pertinent Laboratory Investigations.
|
Baseline Laboratory Tests |
Initial values |
Units |
Reference Range |
|
Sodium |
142 |
mmol/L |
135-145 |
|
Potassium |
3.4 |
mmol/L |
3.5-5.0 |
|
Calcium |
1.86 |
mmol/L |
2.2-2.55 |
|
Albumin |
31 |
g/L |
32-35 |
|
Glucose |
6.7 |
mmol/L |
3.4-11.0 |
|
Urea |
6.9 |
mmol/L |
<11.9 |
|
Creatinine |
67 |
umol/L |
55-100 |
|
Leukocytes |
6.3 |
x109/L |
10-Apr |
|
Hemoglobin |
49 |
g/L |
115-160 |
|
Platelets |
263 |
x109/L |
150-400 |
|
ALT |
16 |
U/L |
<32 |
|
ALP |
75 |
U/L |
35-104 |
|
Total Bilirubin |
7 |
umol/L |
<21 |
|
Haptoglobin |
1.36 |
g/L |
0.3-2.0 |
|
LDH |
240 |
U/L |
<214 |
|
Ferritin |
13.9 |
ug/L |
11-56 |
|
INR |
1 |
|
0.9-1.1 |
|
aPTT |
64 |
seconds |
20-29 |
|
FVIII:C Activity |
<0.01 |
IU/mL |
<0.01 |
|
FVIII Inhibitor Units |
28.8 |
Bethesda Unit |
0 |
|
FIX Activity |
1.57 |
IU/mL |
0.5-2.0 |
|
FXI Activity |
0.75 |
IU/mL |
0.5-2.0 |
|
VWF:Ag |
0.12 |
IU/mL |
0.5-2.0 |
|
VWF:GPIbM Activity |
<0.15 |
IU/mL |
0.48-1.73 |
|
Referred Laboratory Tests VWFpp |
1.30 |
IU/mL |
0.56-1.4 |
|
VWF:GPIbM Activity |
0.13 |
IU/mL |
0.52-1.8 |
|
VWF multimers |
Faint multimeric bands but no apparent preferential loss of the high molecular weight multimers. |
||
|
Other Tests |
|
|
|
|
Rheumatoid factor |
<10 |
IU/mL |
<14 |
|
pANCA |
3 |
RU/mL |
<19 |
|
cANCA |
<2 |
RU/mL |
<19 |
|
ANA |
<1:80 |
|
<1:80 |
|
SPEP |
No monoclonal spike or abnormal bands |
||
|
Serum immunofixation |
No monoclonal protein detected |
||
An urgent upper endoscopy revealed a small, non-bleeding gastric erosion, while colonoscopy identified numerous bleeding angioectasias that required argon plasma coagulation. Hematology was consulted for an isolated prolonged Activated Partial Thromboplastin Time (APTT) of 64 seconds (reference: 20-29 seconds). Testing confirmed a diagnosis of AHA, with FVIII activity <0.01 IU/mL (reference: 0.50-2.00 IU/mL) and a FVIII inhibitor level of 28.8 BU (reference: <0.6 BU). Unexpectedly, both VWF antigen (VWF:Ag) and VWF activity (VWF:GPIbM) were found to be low at 0.12 IU/mL (reference: 0.50- 2.00 IU/mL) and <0.15 IU/mL (reference: 0.48-1.73 IU/ mL), respectively. The VWF:GPIbM/VWF:Ag ratio was not calculable due to the VWF activity being below the detection limit. These results confirm the diagnosis of AHA along with von Willebrand Factor deficiency. With no personal or family history of a bleeding disorder, we considered the possibility of both AHA and AVWS. To manage the active bleeding, IV susoctocog alfa (Obizur®) was administered. An initial dose of 100U/kg IV resulted in a FVIII activity recovery to 0.84 IU/mL. However, due to the low levels of von Willebrand Factor, the estimated half-life of Obizur® was short, measuring approximately two hours. Given the absence of clear causes for AVWS on physical exam and investigations, we suspected immunemediated AVWS. Therefore, we administered Intravenous Immunoglobulins (IVIG) at a dose of 1g/kg and initiated oral cyclophosphamide and prednisone, each at 1mg/kg. Approximately 24 hours after IVIG administration, VWF testing showed rapid improvements in both VWF antigen and activity, suggesting a positive response to immunomodulation with IVIG (Figure 1). By 48 hours after the initial IVIG dose, VWF levels had normalized, which led to an improvement in the half-life of Obizur® to approximately 10 hours in the presence of endogenous VWF, which protects FVIII from premature clearance.
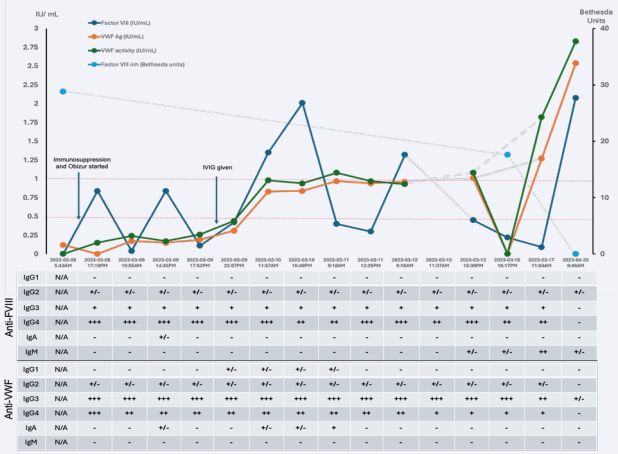
Figure 1 Timeline of the Patient’s Clinical Course and Associated Diagnostic Investigations shown are FVIII levels (Blue), VWF Ag levels (Orange), VWF activity (Green) and FVIII inhibitor level (Teal) as a function of the patient’s clinical timeline. ELISA assessing for presence of autoantibodies are correlated to the clinical dates. - : ODpatient < mean ODCTLneg+3Std Dev +/- : ODpatient > mean ODCTLneg+3Std Dev; ODpatient mean ODCTLneg+3Std Dev; ODpatient ≥ 0.1 but mean ODCTLneg+3Std Dev; ODpatient ≥ 0.4 but mean ODCTLneg+3Std Dev; ODpatient ≥ 0.8.
The patient responded positively to treatment, with no further gastrointestinal bleeding and was discharged after 12 days in the hospital. During her stay, she received transfusions of 3 units of Packed Red Blood Cells (pRBCs) and a total of 91,500 units of Obizur® (approximately 817 U/kg) over ten days of exposure. Further workup with an echocardiogram showed aortic sclerosis without stenosis, with a negative bubble study.
The CT body and PET-CT were not suggestive of malignancy. Additional laboratory testing was sent to a reference laboratory to review VWF Propeptide Antigen (VWFpp) and VWF Inhibitor Screen. These samples were drawn about 30 hours after admission and post-dose IVIG, with corresponding local laboratory testing showing VWF:Ag of 0.15 IU/mL and VWF:GPIbM of 0.17 IU/mL. The reference laboratory reported a VWF:GPIbM of 0.13 IU/mL and a VWFpp of 1.3 IU/mL. The ratio of VWFpp/VWF GP1bM activity was elevated at 10, suggesting increased clearance. The VWF inhibitor screen with the VWF GPIbM mixing study was negative. However, antibodies causing clearance without inhibition of function will not be detected by the assay.
We retrospectively tested stored serial plasma samples using Enzyme-Linked Immunosorbent Assay (ELISA), which revealed the presence of polyclonal anti-FVIII antibodies (predominantly IgG-4 subtype) and anti-VWF antibodies (predominantly IgG-3 subtype) . There was a clear inverse relationship between the optical density on ELISA and the improving VWF profile and FVIII activity, consistent with a response to therapy. This finding confirms the immune-mediated AVWS and AHA diagnoses. Further ELISA testing with different VWF concentrates (Humate-P®, Wilate®, Vonvendi®) showed the binding of anti-VWF IgG antibodies to all concentrates. Notably, Vonvendi® is a recombinant VWF concentrate that lacks FVIII, whereas Humate-P® and Wilate® contain FVIII. Given the distinct polyclonal IgG patterns and the specificity of the IgG autoantibodies toward Vonvendi®, we conclude that two distinct autoantibodies are responsible for the diagnoses of AHA and immune-mediated AVWS. At 4 months, the patient presented with gingival bleeding, requiring hospital admission.
Further testing confirmed a relapse of AHA without AVWS relapse. FVIII activity was 0.08 IU/mL, with a FVIII inhibitor level of 8.8 BU, while the VWF profile remained within normal (VWF:Ag 1.21 IU/mL, VWF:GPIbM 1.12 IU/mL). Rechallenge with prednisone monotherapy led to a second complete response. At 7 months, the patient experienced a partial relapse of AHA, discovered incidentally when she presented with Escherichia coli bacteremia and an isolated prolonged aPTT of 34 seconds. FVIII activity was mildly reduced at 0.37 IU/mL, a significant drop from 2.04 IU/mL measured three weeks earlier. The VWF
profile was elevated (VWF:Ac 2.82 IU/mL, VWF:GPIbM 2.55 IU/mL), reflecting its role as an acute phase reactant. Immunosuppressive therapy with Rituximab (375 mg/m² IV weekly for 4 weeks) and a short course of prednisone was administered. It is likely that early identification of the relapse prevented a full immune attack that could have further inhibited FVIII activity and led to bleeding. At her last follow-up, 15 months after her initial diagnosis, the patient remained in remission, with normalization of both FVIII activity and her VWF profile.
DISCUSSION
Concomitant AHA and immune-mediated AVWS are exceedingly rare, with only one other case reported in the literature [7]. Given that FVIII pharmacokinetics depends on VWF, we recommend routine assessment of VWF when evaluating patients with isolated prolonged aPTT for AHA, as this can have significant therapeutic implications. In this case, the low VWF levels led to a shortened half-life (~2 hours) of Obizur®, necessitating more frequent dosing initially.
This improved to 10 hours as VWF recovered, causing significant variations in FVIII activity peaks and troughs. Our hemostasis lab’s restricted hours contributed to delays in testing and treatment adjustments, highlighting the critical importance of adequate hemostatic laboratory support, especially during active bleeds to assist with Obizur® dose adjustment; however, access to specialized hemostatic laboratory testing can be challenging [8]. Plasma-derived concentrates containing both FVIII and VWF (e.g., Humate P® or Wilate®) were not attempted in this case due to the rapid resolution of the immunemediated AVWS after IVIG. Whether the patient would have responded to VWF concentrate remains uncertain. Theoretically, the antibodies to VWF could lead to increased VWF clearance, and while the patient might initially respond to VWF concentrate, this response would likely be unsustainable if the antibody persists and continues to cause increased clearance. It is important to note that FVIII replacement therapy with plasma-derived FVIII has limited efficacy when the FVIII inhibitor titer exceeds 5 BU [9].
FVIII inhibitors were detected with the NijmegenBethesda Assay [10], while a modified Bethesda method assessed VWF activity inhibition [11]. These tests lack sensitivity for detecting anti-VWF antibodies that target VWF clearance. ELISA tests identified polyclonal IgG responsible for both AHA and immune-mediated AVWS, consistent with literature findings that show IgG4 is commonly involved in both disorders [12]. Figure 1 shows increased IgA and IgG1 antibodies, likely related to IVIG exposure. Pathogenic IgG2, IgG3, and IgG4 antibodies exhibited similar signal strength before and immediately after IVIG administration, then decreased over time due to immunosuppressive therapy.
IVIG is the recommended treatment for IgG monoclonal gammopathy associated with AVWS [13]. In this case, serum immunofixation did not demonstrate paraproteinemia. The therapeutic response observed in the immune-mediated AVWS appears to be temporally related to IVIG administration, though an early response to cyclophosphamide and prednisone cannot be excluded. The bispecific antibody, Emicizumab®, may have a therapeutic role in future relapses, as it has demonstrated efficacy in treating AHA and as prophylaxis in severe type 3 VWD [14].
CONCLUSION
This is a rare case of concomitant AHA and immunemediated AVWS, and to our knowledge, it is only the second such case reported in the literature.7 The patient responded well to immunosuppressive therapy with cyclophosphamide and prednisone, with the immunemediated AVWS showing a prompt response to IVIG. Recombinant porcine FVIII (Obizur®) was effectively for acute bleed management. AVWS led to a shortened half-life of Obizur®, emphasizing the need to assess VWF levels and ensure frequent laboratory monitoring with specialized hemostasis support when diagnosing and managing AHA.
Conflict of Interest
R.Y. and M.B. have no competing interests to disclose. C.P. has received the LRCP Catalyst Grant from London health Sciences Foundation paid to his institution. C.P. has received paid honoraria from Roche, Abbvie, Sanofi, CSL, BeiGene, AstraZeneca, EusaPharma, FORUS Therapeutics, Bayer, Octapharma, Johnson & Johnson and Pfizer. P.J. has received a research grant paid to her institution from Bayer. She has Royalties from Uptodate paid to her institution. She has received consulting fees from Band/ Guardian, Star/Vega, Roche and BioMarin. P.J. has received paid honorarium from Bayer.
Patient Consent: Patient has given informed consent for this case report.
Author Contributions: R.Y. and C.P. provided direct patient care. R.Y. wrote the manuscript and created figures which was critically reviewed by all authors. M.B.,P.J., and A.B. provided additional ELISA testing of patient bloodwork. All authors contributed to data interpretation.
Funding: This case report did not receive any specific grants from public, commercial or non-profit sectors.
References
- Kruse-Jarres R, Kempton CL, Baudo F, Collins PW, Knoebl P, Leissinger CA, et al. Acquired hemophilia a: updated review of evidence and treatment guidance. Am J Hematol. 2017; 92: 695-705.
- Knoebl P, Marco P, Baudo F, Collins P, Huth-Kühne A, Nemes L, et al. EACH2 registry contributors. demographic and clinical data in acquired hemophilia a: Results from the european acquired haemophilia registry (each2). J Thromb Haemost. 2012; 10: 622-631.
- Franchini M, Mannucci PM. Alloantibodies in von willebrand disease.Semin Thromb Hemost. 2018; 44: 590-594.
- Mehta R, Athar M, Girgis S, Hassan A, Becker RC. Acquired Von Willebrand Syndrome (AVWS) in cardiovascular disease: a state of the art review for clinicians. J Thromb Thrombolysis. 2019; 48: 14- 26.
- Franchini M, Mannucci PM. Acquired von Willebrand syndrome: Focused for hematologists. Haematologica. 2020; 105: 2032-2037.
- Kumar S, Pruthi RK, Nichols WL. Acquired von Willebrand’s syndrome: A single institution experience. Am J Hematol. 2003; 72: 243-247.
- Dicke C, Holstein K, Schneppenheim S, Dittmer R, Schneppenheim R, Bokemeyer C, et al. Acquired hemophilia A and von Willebrand syndrome in a patient with late-onset systemic lupus erythematosus. Exp Hematol Oncol. 2014; 20: 3-21.
- Tripodi A, Santoro RC, Testa S, Molinari AC, Bernardini S, Golato M, et al. Position paper on laboratory testing for patients with haemophilia. A consensus document from SISET, AICE, SIBioC and SIPMeL. Blood Transfus. 2019; 17: 229-236.
- Srivastava A, Santagostino E, Dougall A, Kitchen S, Sutherland M, Pipe SW, et al. WFH Guidelines for the Management of Hemophilia panelists and co-authors. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020; 26: 1-158.
- Castellone DD, Adcock DM. Factor VIII Activity and Inhibitor Assays in the Diagnosis and Treatment of Hemophilia A. Semin Thromb Hemost. 2017; 43: 320-330.
- Miller CH. Monitoring of von Willebrand factor inhibitors in patients with type 3 von Willebrand disease using a quantitative assay. Haemophilia. 2021; 27: 823-829.
- James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebranddisease. Blood. 2013; 122: 636-640.
- Franchini M, Mannucci PM. Acquired von Willebrand syndrome: focused for hematologists. Haematologica. 2020; 105: 2032-2037.
- Thomas VM, Abou-Ismail MY, Lim MY. Off-label use of emicizumab in persons with acquired haemophilia A and von Willebrand disease: A scoping review of the literature. Haemophilia. 2022; 28: 4-17.









































































































































{kind=link}