Variants in TBCK Cause Global Developmental Delay, Dysmorphism, Hypotonia
- 1. Department of Clinical Genetic and Metabolic Genetics, King Saud Medical Hospital, Saudi Arabia
- 2. Riyadh Regional Laboratory, Department of Biochemistry, King Saud Medical Hospital, Saudi Arabia
Abstract
Background: A protein kinase domain, a Rhodanase-like domain, and the Tre-2/Bub2/Cdc16 (TBC) domain are all encoded by the TBCK gene. By modulating the mammalian target of rapamycin (mTOR) signaling pathway, the encoded protein is hypothesized to play a role in actin organization, cell growth, and cell proliferation. Has a role in the organization of the actin cytoskeleton. This protein may also play a role in the transcriptional control of mTOR complex components. And it’s found practically everywhere, including the kidneys, intestines, and other organs.
Methods: The genetic etiology of facial dysmorphic disorder was investigated using whole exome sequencing (WES). Which was requested for two related cousin’s probands have developmental delays and hypotonia?
Results: Splice site variants were found in both patients with an apparently homozygous for splice variants in the TBCK gene, according to WES.
Conclusion: TBC1 domain-containing kinase (TBCK) homozygous mutation is closely linked to a severe global developmental delay. Hypotonia and seizure disorder, as well as a distinctive facial appearance and early death, we present two cousins patients with developmental delay, facial dysmorphism, and hypotonia as a result of TBCK.
Keywords
Global developmental delay; TBCK, Mutations; Whole-exome sequencing
Citation
Alotaibi M, Aloulou SM (2022) Variants in TBCK Cause Global Developmental Delay, Dysmorphism, Hypotonia. JSM Clin Case Rep 10(1): 1197
INTRODUCTION
TBCJ (TBC1 domain containing kinase) is a member of the RabGAP family and can be found on chromosome 4q24. Tre-2, BUB2p, and Cdc16p are three proteins found in the TBC domain. Involved in the regulation of cell proliferation and growth as well as the modulation of mTOR signaling and the expression of mTOR complex components [1]. The collier/olfactory-1/EBF (COE) family of transcription factors is required for the development and differentiation of various cell types across species, from planarians to humans [2-4]. These proteins have been proven to be functional domains of Rab GAP, which could catalyze GTP hydrolysis of Rab GTPase via a dual-finger mechanism. COE proteins share a conserved amino-terminal DNA-binding domain with a unique zinc-finger binding motif, an immunogolubulin Ig-like, plexins, transcription factors (IPT/TIG) domain, and an unusual helix–loop–helix domain with limited sequence similarity to other protein families.
TBCK mutations have previously been identified in children with hypotonia, infantile psychomotor delay, and dysmorphic [5-7].
Two siblings of consanguineous Saudi parents [7] were found to have a homozygous splice site mutation implicating the TBCK gene in molecular genetic research in a large cohort of 143 Saudi families with various neurodevelopmental disorders. Whole-exome sequencing revealed homozygous mutations in the TBCK gene in five patients [5], two of whom were sisters from different ethnic backgrounds (Lebanese, Egyptian, and Puerto Rican) and the other with a syndromic infantile encephalopathy characterized by severe developmental disability, brain atrophy, seizures, central respiratory failure, and facial dysmorphism.
They discovered 13 individuals from nine separate families that have biallelic mutations in TBCK and have unusual facial traits, intellectual impairments, and hypotonia in a previous research [6]. Confirmed Whole-exome sequencing and Sanger sequencing validated the TBCK mutations. In patient cells with a splicing site mutation, they discovered undetectable amounts of the TBCK protein, as well as decreased phosphorylation of phosphoribosomal protein S6 in investigations of patient cells with a distinct frame shift mutation.
The clinical and molecular findings of two related patients with global developmental delay, dysmorphism, and hypotonia are presented in this work. Variants in TBCK were discovered using WES.
CLINICAL PRESENTATION
Patient 1
He is a 3-year-old Saudi boy who was diagnosed with dysmorphic traits, significant hypotonia, and global developmental delay at the age of two. Her parents were consanguineous and had had one abortion in the past. At the age of two, he sat unaided and did not walk. His weight was 13.5 kg (0.40 SD), his height was 92 cm (0.33 SD), and his head circumference was 48 cm. He had a history of generalized tonic/ clonic seizures around the age of 3 months, which were effectively managed with Levetiracetam (0.35 SD). Slanted forehead, epicanthal fold, bitemporal constriction, bulbous nose, thick mouth, macroglossia, and large ears were among his features. He had severe hypotonia and had little to no reflexes. Hearing was normal, and the ophthalmology check was uneventful. On G-tube feeding, he had a history of difficulty swallowing. Recurrent admissions for chest infection since the age of two months. A brain MRI revealed bilateral diffuse loss of normal cerebral white matter bulk, as well as a minimal irregular lateral outline of the lateral ventricles, which was more pronounced at the posterior horns, and residual brain parenchyma with high T2 and FLAIR signal intensities and no diffusion restriction. At the age of two, an EEG revealed epileptiform discharges. Previous genetic tests, such as karyotype, chromosomal microarray, and metabolic screening (tandem MS, urine organic acids), all showed normal CK and normal karyotype. Whole Exome Sequencing has been discovered. c.1897+1G> seems to be homozygous. In this patient, a pathogenic mutation in the TBCK gene was discovered. This variation has been linked to disease in the past (PMID: 25558065, 27040691, ClinVar ID: 183338). Hypotonia, infantile, with psychomotor retardation and distinctive facies 3 [OMIM:616900], an autosomal recessive neurodevelopmental condition, is caused by TBCK defects. Characterized by profound developmental disability, intellectual disability and severe hypotonia.
Patient 2
She is a 2-year-old Saudi girl who is the only child of two healthy Saudi parents who are both suffering from severe hypotonia. She had severe developmental delays, blindness, macrocephaly, and dysmorphic characteristics such as brachycephaly, epicanthal folds, bitemporal constriction deep set eyes, bulbous nose, anteverted nares, and tented upper lip. Treatment for uncontrollable focal seizures and the insertion of a feeding tube. She was admitted to pediatric intensive care multiple times due to severe chest infections; at the age of two, she became ventilator dependent and died of cardiac arrest.
Periventricular leukomalacia is seen on MRI, and white matter alterations corroborate the diagnosis. leukodystrophy illustration The first workup included metabolic tests, urine GCMS, quantitive amino acid testing, CSF amino acid testing, and neurotransmitter testing, all of which were normal. Whole exome sequencing has been discovered. The TBCK variation c.1897+1G>A is projected to disrupt the donor splice site, which is highly conserved. According to HGMD Professional 2020.1, Alazami et al. 2015 (PMID: 25558065) [7], Bhoj et al. 2016 [6], identified this variant as causing global developmental delay, epilepsy, dysmorphism, hypotonia, delayed reflexes, and VSD (PMID: 27040691). This variation is classified as probable pathogenic by ClinVar (research, Variation ID: 183338). According to the guidelines of CENTOGENE and ACMG, it is classed as pathogenic (class 1).
Genetic
studies Peripheral blood samples (5 ml) were collected in EDTA tubes from the index case and parent’s written informed consent was obtained before the samples’ collection. Whole exome sequencing (WES): for the paired-end pre-capture library procedure, genome DNA is fragmented by sonicating genomic DNA and ligating to the Illumina multiplexing PE adapters. The adapter-ligated DNA is then PCR amplified using primers with sequencing barcodes (indexes). For target enrichment/exome capture procedure, the pre-capture library is enriched by hybridizing to biotin labeled VCRome 2.1 in-solution exome probes. Additional probes for over 3600 Mendelian disease genes were also included in the capture in order to improve the exome coverage.
It revealed the presence of a homozygous of the TBCK variant c.1897+1G>A is predicted to disrupt the highly conserved donor splice site (Table 1).
|
Table 1: Identified TBCK gene mutation. |
|||||||
|
Disease |
Inheritance pattern |
Gene |
Position |
isoform |
Nucleotide |
Zygosity |
TYPE AND CLASSIFICATION |
|
Infantile hypotonia with psychomotor retardation and characteristic facies 3 [MIM:616900] |
AR |
TBCK |
chr4: 10711587 |
NM_001163435. 1 |
1897+1G>A |
HOM |
Splicing Pathogenic (class 1) |
According to HGMD Professional 2020.1, this variant has previously been described as disease causing for Global developmental delay, epilepsy, dysmorphism, hypotonia, delayed reflexes, and VSD by Alazami et al., 2015 (PMID: 25558065) [7], Bhoj et al., 2016 (PMID: 27040691) [6]. ClinVar lists this variant as likely pathogenic (research, Variation ID: 183338). It is classified as pathogenic (class 1) according to the recommendations of CENTOGENE and ACMG.
DISCUSSION
Infantile hypotonia with psychomotor retardation and distinctive facies-3 is a severe autosomal recessive neurodevelopmental condition that affects children from birth to early childhood. Most affected individuals have delayed speech, seizures that are often resistant to pharmacological treatment, cortical visual impairment, and coarse facial features with other dysmorphism facial appearances such as bitemporal narrowing, deep-set eyes, high nasal bridge and long philtrum, tented upper lip, macroglossia, and specific radiological brain findings that range from non-specific to a leukodystrophy process in the more severe cases. To date, a large-scale WES project in consanguineous couples has found splicing mutations in TBCK in all cases.
We presented two individuals diagnosed with full exome (TBCK variation c.1897+1G>A) from two separate Saudi families. They all had the clinical phenotype of infantile hypotonia with psychomotor retardation and typical facies 3 illnesses, which was characterized by craniofacial dysmorphic characteristics such as a sloping forehead, macrocephaly, bulbous nose, and coarse features. Patients with extensive brain atrophy, enlarged ventricles, and periventricular white matter anomalies were found to have neurological deficits, including severe hypotonia, decreased reflexes, and varying degrees of delayed psychomotor development. Vision problems have not been detected in our patients.
Variants in disease genes that are linked to clinical phenotype (Table 2) c.1897+1G> seems to be homozygous. In our patients, a pathogenic variation in the TBCK gene was discovered, which is projected to disrupt the donor splice site, which is highly conserved.
|
Table 2: Phenotype-Genotype comparison of 5 patients from reference 5 with our patients. |
|||||||
|
Family |
1 |
2 |
3 |
4 |
5 |
Our case 1 |
Our case 2 |
|
Age |
14 years |
4 years |
10 years |
24 months |
14 years |
3 years |
2 years |
|
Sex |
male |
female |
Female |
male |
male |
male |
female |
|
Consanguine- ous |
no |
yes |
yes |
yes |
no |
yes |
yes |
|
Ethnicity |
Puerto Rica |
Lebanese |
lebanese |
Egyptian |
Puerto Rica |
Saudi |
Saudi |
|
Developmen- tal |
Severely de- layed |
profound |
profound |
profound |
profound |
profound |
profound |
|
Seizure |
+ |
+ |
+ |
No |
+ |
+ |
+ |
|
Hypotonia |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
Vision |
Cataract, blindness |
Vision impairment |
Vision impairment |
Bilateral optic atrophy |
Vision impair- ment |
No vision impair- ment |
Vision impair- ment |
|
MRI brain |
ventricu- lomegaly decreased white-matter volume and thin corpus callosum |
ventriculomegaly, thin but complete corpus callosum, and mild cerebellar vermis hypoplasia |
ventriculomegaly, thin but complete corpus callosum, and mild cerebellar vermis hypoplasia |
ventriculom- egaly, absent corpus callo- sum, and mild cerebellar vermis hypo- plasia |
ventriculom- egaly, thin cor- pus callosum, and mild cer- ebellar vermis hypoplasia |
bilateral diffuse loss of the normal cer- ebral white matter |
periventricular leukomalacia, and white-matter changes |
|
c.376C>T (p.Arg126 |
c.1363A>T (p.Lys455 |
c.1363A>T (p.Lys455 |
c.1532G>A (p.Arg511 |
c.376C>T (p.Arg126 |
c.1897+1G>A |
c.1897+1G>A |
|
This variation has been linked to disease in the past (PMID: 25558065, 27040691, ClinVar ID: 183338), In two Saudi siblings, Alazami et al, 2015 [7] discovered a homozygous splice site mutation in the TBCK gene. Global developmental delay, epilepsy, dysmorphism, hypotonia, and VSD were all present (Figure 1).
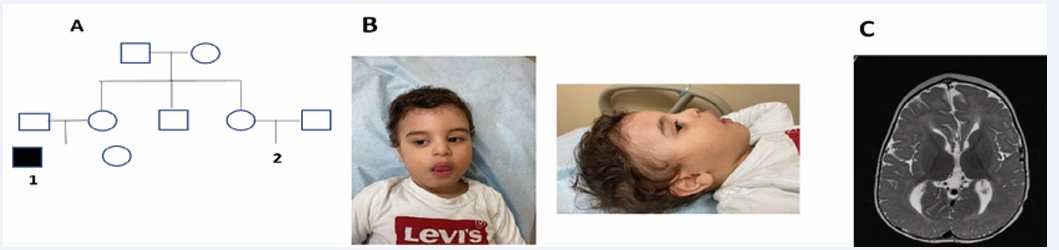
Figure 1 A) pedigree of the family affected, B) photographs of case 1 (slanted forehead, epicanthal fold, bitemporal constriction, bulbous nose, thick mouth, and large ears), C) Mri brain of case 1 showed (bilateral diffuse loss of normal cerebral white matter bulk, minimal irregular lateral outline of the lateral ventricles and residual brain parenchyma with high T2 and FLAIR signal intensities and no diffusion restriction.).
(Bhoj et al, 2016) [6] documented 13 people with pathogenic homozygous TBCK mutations who had distinctive facial dysmorphism, intellectual impairments, and hypotonia from various families and ethnicities.
He also concluded that increasing mTOR signaling with the addition of leucine to the culture media in afflicted cells was a therapeutic strategy. This could be employed as a therapeutic effect in these individuals, as a deficiency in TBCK in cells causes cell proliferation to be disrupted and Ser235/ phosphorylation to be reduced. Guerreiro et al. 2016 [8], reported a novel 4-bp deletion in the gene TBCK, which was considered the deletion mutation within exon 11 and generates a premature stop codon, in three siblings with severe hypotonia, global developmental delay, and distinctive facial traits. One of the fundamental causes of developmental delay is the TBCK mutation.
Finally, TBCK abnormalities are the source of infantile hypotonia with psychomotor impairment and other symptoms. On neuroimaging, many individuals experience seizures and have brain atrophy, dysgenesis of the corpus callosum, and white matter changes. Some people have facial dysmorphism that isn’t specific. The findings may be in consistent with the phenotype reported.
ACKNOWLEDGEMENTS
We highly appreciate the patient and his parents for consenting and participating in this study.
REFERENCES
- Yueli Liu, Xiaoyi Yan, Tianhua Zhou. 2019. TBCK Influences Cell Proliferation, Cell Size and mTOR Signaling Pathway. PLoS One. 2013; 8: 71349.
- Wen Bu, Li-Kuo Su. Characterization of Functional Domains of Human EB1 Family Proteins. J Biol Chem. 2003; 278: 49721-49731.
- Marina I Siponen, Magdalena Wisniewska, Lari Lehtiö, Ida Johansson, Linda Svensson, Grzegorz Raszewski, et al. Structural Determination of Functional Domains in Early B-cell Factor (EBF) Family of Transcription Factors Reveals Similarities to Rel DNA-binding Proteins and a Novel Dimerization Motif. J Biol Chem. 2010; 285: 25875-25879.
- Martis W Cowles, Kerilyn C Omuro, Brianna N Stanley, Carlo G Quintanilla, Ricardo M Zayas. COE Loss-of-Function Analysis Reveals a Genetic Program Underlying Maintenance and Regeneration of the Nervous System in Planarians. PLoS Genet. 2014; 10: 1004746.
- Jessica X Chong, Viviana Caputo, Ian G Phelps, Lorenzo Stella, Lisa Worgan, Jennifer C Dempsey, et al. Recessive Inactivating Mutations in TBCK, Encoding a Rab GTPase-Activating Protein, Cause Severe Infantile Syndromic Encephalopathy. Am J Hum Genet. 2016; 98: 772- 781.
- Elizabeth J. Bhoj, Dong Li, Margaret Harr, Shimon Edvardson, Orly Elpeleg, Elizabeth Chisholm, et al. Mutations in TBCK, Encoding TBC1-Domain-Containing Kinase, Lead to a Recognizable Syndrome of Intellectual Disability and Hypotonia. Am J Hum Genet. 2016; 98: 782-788.
- Anas M Alazami, Nisha Patel, Hanan E Shamseldin, Shamsa Anazi, Mohammed S Al-Dosari, Fatema Alzahrani, et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole- Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015; 10: 148-161.
- Rita J Guerreiro, Rachel Brown, Donnai Dian, Christian de Goede, Jose Bras, Sara E Mole, et al. Mutation of TBCK causes a rare recessive developmental disorder. Neurol Genet. 2016; 2: e76.









































































































































{kind=link}