Analysis of Beta-Blockers in Environment - A Review
- 1. Laboratoire de Photochimie et d’Analyse, Département de Chimie, Faculté des Sciences et Techniques, Université Cheikh Anta Diop, Dakar, Sénégal
- 2. Laboratoire Géomatériaux et Environnement (LGE), EA 4119, Université Paris-Est Marne-la-Vallée, 5 Boulevard Descartes, Bâtiment IFI, 77454 Marne la Vallée Cedex 2, France
- 3. Equipe des Matériaux, Electrochimie et Photochimie Analytique, Département de Chimie, Université Alioune Diop de Bambey
Abstract
The β-blockers are among the most prescribed drugs for the treatment of cardiovascular diseases in the world. They have been mainly applied to the treatment of hypertension, heart failure, myocardial infarction and cardiac arrhythmias. Also, due to their massive use, β-blocker pharmaceutical residues have been recently detected in environmental waters. Once released into the natural environment by Waste Water Treatment Plants (WWTPs), residues of β-blockers can accumulate in aquatic media and aquatic species. Therefore, a great number of research studies have been carried out on their ecotoxicity for aquatic organisms, and potential risks for human health. The β-blockers are also used as doping agent by athletes and illegally injected to animals in order to reduce their mortality. Therefore, it was essential to develop appropriate analytical procedures to provide reliable data related to their presence and concentrations in various matrices. The present study investigated the development of analytical approaches of β-blockers reported in the literature between 2000 and 2022, and their occurrence in different matrices with also a focus on sample extraction procedures.
Graphical Abstract
KEYWORDS
- β-Blockers; Sample Preparation; Liquid & Gas Chromatography; Spectrofluorimetry; Distribution
CITATION
Gueye C, Cissé L, Diaw PA, Mbaye OMA, Seye MDG, et al. (2025) Analysis of Beta-Blockers in Environment - A Review. JSM Environ Sci Ecol 13(1): 1106.
INTRODUCTION
Cardiovascular diseases constitute one of the leading causes of mortality in the world, representing around 31% of deaths (WHO report, 2017). Due to the high prevalence of these cardiovascular diseases, β-blockers remain among the most prescribed drugs worldwide. They are used in the treatment of hypertension, heart failure, acute myocardial infarction, cardiac arrhythmias, glaucoma, migraine or anxiety [1]. Indeed, β-blockers, also called adrenoreceptor antagonists, act by blocking the release of catecholamines (adrenaline and noradrenaline, which play the role of hormone or neurotransmitter) on β-adrenergic receptors, in order to slow down nerve impulses and heart rate. Thus, β-blockers are increasingly utilized, their consumption having almost doubled between 2000 and 2017 in OECD countries. Indeed, industrialized countries are the major consumers of β-blockers, their types and quantities varying according to the country. For example, β-blockers, such as atenolol, metoprolol and propranolol, are the most consumed (more than 18 tons of atenolol in 2004 in France [2] and about 100 tons per year in Germany [3]. In the United States, β-blockers are in the top one hundred most prescribed pharmaceuticals [4] with more than 89 million prescriptions for metoprolol in 2017 [fourth among most consumed drugs [5]. In China, the annual consumption of metoprolol has increased from about 27.9 kg in 2011 to 63.8 kg in 2015, which can be attributed to the increase of number of hypertensive patients [6]. Also, in the United Arab Emirates, the global consumption of β-blockers has reached more than one million units in 2010 [7]. All the above figures show that the evolution of the world consumption of β-blockers is strongly increasing.
After consumption, pharmaceutical products are excreted either under unchanged form or partially metabolized via urine and feces to end up in wastewaters. These wastewaters are often transferred by sewage networks to Wastewater Treatment Plants (WWTP) where they can undergo different types of treatment. However, several studies have revealed the inefficiency of WWTPs for the total elimination of pharmaceuticals [8,9]. As a result, WWTPs are the main sources of release of pharmaceutical compounds into the environment. Moreover, they can penetrate the soil after sludge spreading or through the reuse of treated wastewater in agriculture. Consequently, these substances are constantly released into the environment, and numerous workers have confirmed the ubiquity of β-blockers in the environment worldwide, at concentrations in the range ng/L to µg/L [10-20]. For example, more than twelve β-blockers have been listed and detected in various environmental matrices on a global scale with concentrations that vary from one country to another, depending on the consumption but also on the type of β-blocker [17-22]. However, few studies have been realized on the contamination of African waters by β-blockers. Nevertheless, high concentrations of pharmaceutical residues have been found in African waters due to the lack of treatment system infrastructure. For instance, atenolol has been found in high concentrations reaching about 39 µg/L in the Umgeni River, South Africa [23] and 3 µg/L in a river in Lagos, Nigeria [24].
Residues of pharmaceuticals can also accumulate in aquatic species [25] and in plants [26]. Their bioaccumulation has been shown in aquatic organisms, such as fish, shellfish and algae, after prolonged exposure [27]. For example, Moreno-González et al., [28] reported the presence of metoprolol at a concentration of 0.7 ng/g of golden mullet (Liza aurata) in Mediterranean sea, and the β-blockers, carazolol and propranolol, have been found in fish at levels of, respectively, 3.8 and 4.2 ng/g (dry weight) [29]. In addition, the presence of β-blockers was determined in animal products (meat, muscle, kidneys, liver and dairy products) [30,31], which could yield potential risks to the ecosystem and human health, and toxicological effects to aquatic organisms [32-36]. Also, Godoy et al [34] have shown that β-blockers, like propranolol, metoprolol and sotalol, had harmful effects on algae and fish. Moreover, it has been demonstrated that two atenolol biotransformation products (ATE-167 and ATE- 117) and one metoprolol biotransformation product were more toxic than the parent compounds. β-blockers and their metabolites are considered as endocrine disruptors because they have the ability to modify the aquatic species hormonal system [37]. β-blockers are biologically active, persistent and accumulate in environmental waters. Therefore, even at low concentrations, they can affect aquatic organisms and the ecosystem. For this reason, β-blockers have been added to the list of emerging contaminants present in the environment, such as pesticides and polycyclic hydrocarbons [38].
In addition to their therapeutic uses, β-blockers have been utilized as doping agents in sport to enhance the athletic performances. Therefore, since 1988 they have been included in the list of doping agents prohibited by the International Olympic Commission in certain competitions [39]. Similarly, in animal husbandry, β-blockers have been often illegally injected into animals during transportation to slaughterhouses in order to reduce anxiety and to prevent the death of animals en route [40]. Therefore, β-blockers accumulate in the edible tissues of animals (muscles, liver, kidneys and milk) which can lead to potential health risks for consumers. Consequently, some countries have set maximum residue limits (MRLs) for these compounds. For example, the MRL for the β-blocker carazolol was set at 25 µg/kg for pig kidney and at 15 µg/kg for bovine kidney [41]. Also, the European Union suggested a maximum residue limit of 1.0 μg/kg for carazolol in milk powder.
Therefore, the analysis of β-blockers in biological fluids, pharmaceutical formulations, food and environmental waters was essential for the following fields: control of therapeutic compliance, intoxication, doping control in sport and ecotoxicity studies. As a result, several analytical methods have been developed for their detection and quantification in different matrices. Liquid chromatography (LC) and gas chromatography (GC) were widely used both in biological fluids and in environmental matrices. However, the GC analysis of β-blockers required derivation steps, which were generally long and tedious. For this reason, GC has been increasingly replaced by LC. In most cases, chromatographic methods were coupled with detection techniques, such as mass spectrometry (MS), tandem mass spectrometry (MS/ MS), UV-VIS spectrometry, DAD spectrophotometry and fluorimetry. Other separation methods, such as capillary electrophoresis have been developed for determining β-blockers in environmental waters [42,43]. Due to their natural fluorescence properties, spectrofluorimetry was also used for analyzing some β-blockers [44-47].
Until now, very few reviews were devoted to the methods of analysis of β-blockers in various matrices. For example, Y?ld?r?m et al. [48] reviewed different analytical methods of β-blockers in several matrices, and Yi et al. [22] provided a review on the distribution of β-blockers, their transformation, their ecotoxicity and their fate in the environment. Therefore, in the present study, we reviewed the different analytical methods used for the determination of β-blockers in the environment and we provided a comprehensive review on their concentration levels in biological fluids, pharmaceutical formulations, and environmental matrices, food of animal origin, biotic species and agro-ecosystem. Our review concerned papers mainly published between 2000 and 2022, covering chromatographic methods and spectrofluorimetric, for multi-residue analysis of β-blockers in different matrices. Indeed, the spectrofluorimetric approach could serve as an alternative method, because it was rather inexpensive, rapid, and very sensitive for the determination of β-blockers. In this review, we described also the different sample extraction procedures as well as the method validation parameters, such as recovery rates, linearity, detection limits and quantification limits. Our objective was to provide an overview of the current state of analytical methods applied to β-blockers, and to better understand their presence in the environment as emerging contaminants.
PHYSICOCHEMICAL PROPERTIES
All β-blockers possess in their chemical structures an aromatic or heteroaromatic ring and an aryloxypropanolamine side chain with an asymmetric carbon. All of these compounds have at least one chiral center, except for timolol, which is marketed as the pure, active S-enantiomer, the other drugs are sold as 50:50 racemic mixtures in the S- and R-forms, which could possess markedly different pharmacodynamic and pharmacokinetic behaviors [49]. Beta-blockers are weak bases with acidity constant (pKa) values greater than 9. Most beta-blockers are soluble in water, but their solubility varies widely. For example, carazolol has a low solubility of 8.52 mg/L, whereas the metoprolol solubility is 16900 mg/L (Table 1).
Table 1: Physicochemical properties of some β-blockers [51,52].
|
β-blocker |
Molecular weight |
Chemical structure |
pKa |
Log P |
Water Solubility (mg/L) |
Excretion in urine (%) |
|
Sotalol |
272.36 |
|
9.43 |
0.24 |
5510 |
75 |
|
Pindolol |
248.33 |
|
9.46 |
1.75 |
860 |
- |
|
Bisoprolol |
325.44 |
|
9.5 |
1.87 |
2240 |
50 |
|
Atenolol |
266.34 |
|
9.3 |
0.16 |
13300 |
40-50 |
|
Propranolol |
259.37 |
|
9.2 |
2.6 |
70 |
1-4 |
|
Metoprolol |
267.18 |
|
9.7 |
1.69 |
16900 |
5-10 |
|
Carazolol |
298 |
|
9.6 |
3.59 |
8.52 |
- |







The value of log P, represents the lipophilicity of a compound, that is to say the strength of the interaction of a molecule with lipids. This property makes it possible to estimate the ability of a molecule to penetrate plant and animal cells. The smaller the log P value will be, the more affinity the molecule will have with water. A log P value greater than 5 indicates that the substance is lipophilic, and therefore has an important potential for bioaccumulation [50]. For many β-blockers, their partition coefficient (log P) is very low, which indicates that these compounds are hydrophilic and do not accumulate very much in organic matter. Based on pharmacokinetic data, the human excretion rates of β-blockers in their original form can vary from 4% for propranolol to 75% for sotalol.
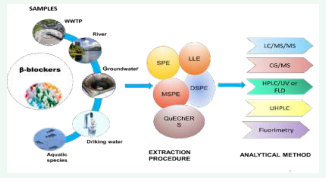
EXTRACTION PROCEDURES OF BETA-BLOCKERS FROM VARIOUS SAMPLES
Due to the complexity of environmental and biological matrices, the analysis of traces of β-blockers in these media required efficient pretreatment and extraction procedures to minimize or eliminate interferences. Therefore, different sample pre-treatments, such as the Liquid-Liquid Extraction (LLE), solid phase extraction (SPE), Solid-Phase Microextraction (SPME), Liquid-Phase Microextraction (LPME), were used for the extraction of β-blockers in various samples.
The LLE extraction procedure has long been used for the preparation and purification of β-blockers in biological samples [53-55]. The LLE extraction procedure consisted in a separation, i.e. an unequal distribution of solutes between two immiscible liquid phases. The most common solvents used for LLE of β-blockers included hexane, benzene, methanol, acetonitrile, ethyl ether, butyl acetate, dichloromethane and various solvent mixtures. This technique was successfully utilized to extract β-blockers with recovery values above 80% in most studies [56]. Thus, the LLE had indisputable advantages, such as simplicity, reliability and adaptability to a wide variety of sample types and analytes. However, it suffered from certain drawbacks, including a long duration and the use of large quantities of organic solvents, generally toxic to the environment. As a result, LLE was increasingly being replaced by SPE. The SPE procedure implied loading a solution onto a solid support (usually a cartridge containing the sorbent), capable of retaining the target analytes, removing unwanted components, and washing/ eluting the desired analytes with another solvent into a collection tube [57].
There is a wide range of sorbents commercialized in the market, such as Hydrophilic-Lipophilic Balanced (HLB), Mixed Phase-Cation Exchange (MCX) or Mixed Phase- Anion Exchange (MAX) and C18 sorbent. The use of Oasis- HLB cartridge SPE has been widely reported in numerous studies for the extraction of β-blockers in various matrices [51]. In order to evaluate the performances of the SPE extraction, different types of sorbents have been evaluated, as well as the effect of the pH and of the solvent used as eluent during the extraction of β-blockers [58- 60]. For example, Gros et al. (2006) tested different types of SPE cartridges, such as Oasis-MCX, C18 and Oasis-HLB for the analysis of different pharmaceutical compounds, including β-blockers. The literature results showed that the best recovery rates were obtained with Oasis-HLB sorbents at values greater than 60% for the β-blockers, as well as the other compounds (acidic and neutral), except sotalol, compared to the cartridges. This sorbent, with the combination of the hydrophilic–lipophilic polymer, can extract acidic, neutral and basic analytes at a wide range of pH, including neutral pH.
Highly selective sorbents, such as Molecularly Imprinted Polymers (MIPs), have been also employed to improve analyte recovery. Thus, sorbents based on MIPs were used for the determination of eight β-blockers in wastewater and surface water [61]. Indeed, MIPs are synthesized materials that have a high affinity and a specific recognition capacity for target molecules. In this study, the extraction performances of MIPs were compared to those of Oasis HLB. Similar recovery rates, comprised between 50 and 110 %, were obtained in both cases. Moreover, due to its specificity for the target analytes, the MIP procedure provided lower LODs than those obtained with Oasis- HLB. The advantages of these MIP-based sorbents were a high selectivity and satisfactory recoveries of β-blockers. However, their high cost prevented their use in routine analyses.
Basan and Yar?mkaya [44]. Developed a new SPE- spectrofluorimetric procedure, using poly-hydrogel (acrylic acid-ethylene glycol dimethyl-acrylate) as adsorbent for the extraction of atenolol in urine. Several factors affecting atenolol extraction efficiency, such as the type and volume of washing solvent, and the eluting solvent were optimized. High mean recovery (95.4%) and low RSD values (3.8%) were obtained for spiked atenolol in human urine. However, SPE extraction of atenolol from urine by MIPs gave less satisfactory recoveries, in the range 74.5-75.3% [45]. A comparison study of eight online SPE cartridges for the determination of five β-blockers and two β-agonists was also performed by Caban et al., [62] in environmental waters. The best recoveries of β-blockers were confirmed with the Polystyrene-divinylbenzene- Nvinylpyrrolidone copolymers (PS-DVB-VP), Strata-X and Oasis-HLB cartridges. The advantages of online SPE laid on the use of low sample volumes, full automation, easy application and high cost-effectiveness.
The effect of sample pH on the extraction efficiency was widely investigated. For example, Vieno et al. [63] optimized the effect of sample pH over the range of pH 4-10. In the case of most β-blockers, the pH value did not impact significantly the recovery value, with the exception of atenolol and sotalol, for which the recovery values increased from less than 10% to more than 60% for a pH increase from 4 to 10. These results were confirmed by Nikolai et al. [64], who showed that, at low pH values, the recovery rate of certain β-blockers was slow, with values of 26% and 49%, respectively, for metoprolol and atenolol. Therefore, the extraction of β-blockers by SPE on an HLB cartridge was often carried out without adjusting the pH of the sample. Moreover, MacLeod et al. [65] extracted the enantiomers of β-blockers, using the SPE procedure. This study showed that the HLB cartridge SPE extraction process did not favor one enantiomer over another, and that there was no difference in recovery rates between enantiomers for most analytes. However, differences were noted in the recovery efficiency between the stereoisomers of nadolol and sotalol in wastewater samples. For example, in the case of sotalol, the recovery rates of the E1 enantiomer (90%) were higher than those of E2 (64 %).
Strong polymer mixed phase-cation exchange (Oasis MCX) SPE has been used for the extraction of β-blockers in environmental waters Lee et al. [49]. MCX sorbents were designed for the extraction of basic and neutral compounds and, generally, provided good recovery for β-blockers. The results of this study indicated recovery rates greater than 80% for the β-blockers, except for pindolol which was not extracted in these samples. Similarly, YongNing et al. [66] extracted five β-blockers from pork and chicken muscle using a MCX extraction cartridge (SPE). A mass of 5.0 g of the homogenized muscle sample was weighed and placed in a 50-mL polypropylene centrifuge tube, and 10 mL of a 5% trichloroacetic acid solution was added to the tube. The mixture was vortexed for 30 sec, then sonicated at 80°C for 30 min. The extract pH was adjusted between 2 to 10, before the extract was applied to the cartridge. It was found that a pH 4.0 value gave the best recoveries for almost all analytes. The recoveries of each compound in the spiked samples at three levels 5, 10, 20 µg/kg were in the range of 47.3 - 123.7%, and the relative standard deviations were in the range of 3.2-25.7%.
In general, the SPE procedure was efficient, economic and environmental-friendly, because it required a smaller amount of solvent than LLE. However, it requested several steps, and losses of analysts were observed during the processes. Therefore, alternative microextraction techniques, such as SPME and LPME, were used for the extraction of beta-blockers in biological and environmental samples. These procedures had advantages over LLE and SPE in terms of reduced amounts of solvent, ease of sample handling, and speed. A two-phase Hollow Fiber protected Liquid-Phase Micro-Extraction (HF-LPME) technique was developed for the analysis of six β-blockers in natural waters by Li et al. [16]. LPME was a miniaturized LLE extraction procedure in which small volumes of solvent were sufficient to concentrate analytes. Several parameters including the type of extraction, solvent and pH, which could affect the extraction efficiency were optimized. The best recovery rates, ranging from 96.7 to 101.1% were obtained in heptanol, compared to hexane and toluene. Recently, a new extraction procedure based on liquid-liquid microextraction, using a deep eutectic solvent, was used for the extraction of three β-blockers, including atenolol, propranolol and metoprolol, from plasma samples.
Jakubus, et al [67] reported a dispersive Solid Phase Extraction (dSPE) process to extract six β-blockers from environmental waters. Multi-walled carbon nanotubes (MWCNTs) were synthesized as extraction sorbent. The dSPE process was also used to extract 14 β-blocker residues from pig and beef kidneys [68]. Since these samples were solids, a pre-treatment process was carried out by centrifugation before extraction. Recovery rates for this method were between 67 and 93%, and the results showed that the dSPE method was easier and faster than conventional SPE.
Parrilla Vázquez, et al. [69] developed an ultrasound- assisted ion dispersive liquid-liquid microextraction (US-IL-DLLME) procedure for the extraction of nine compounds including three beta-blockers (metoprolol, bisoprolol, bétaxolol), in wastewater. High recovery values in the range of 95-103% were found in water samples for all beta-blockers under study.
A Pressurized Liquid Extraction (PLE) process was used by Ramil et al. [3] for the extraction of sediment samples, with recoveries ranging from 89% to 102%. PLE was a simple and comprehensive extraction process that allowed to obtain quantitative recoveries with minimal effort on method development.
Another on-line extraction technique, based on a Turbo-Flow Column (TFC), previously applied as a cleaning technique for biological samples, such as serum, was used for the extraction of environmental samples in the analysis of fifty eight pharmaceuticals [70]. The method consisted to directly inject a small water sample volume into an on-line system including a Turbo-Flow chromatograph for cleaning, followed by liquid chromatography-mass spectrometry and electrospray (TFC-LC-ESI-MS/MS). The best recovery rates for the nine β-blockers ranged from 85.8 to 116% for groundwater, and from 65 to 120% for surface water. Compared to the previous study of López-Serna et al. [71], TFC was found to have better performances than on-line SPE for the extraction of the same compounds in natural waters. However, the TFC technology was unsuitable for the extraction of more complex samples, such as raw water and treated wastewater. Shang et al. [72] reported a new method for the determination of eleven β-blockers in food, by using the Quick, Simple, Cheap, Efficient, Robust and Safe (QuEchERS) extraction approach. QuEChERS was widely a promising technique that could replace SPE cartridges. In this method, several parameters, such as the type and amount of adsorbent and the adsorption time, were investigated. The results indicated that the PSA and MgSO4 adsorbents had the best recovery rate values.
Thus, a combination of 50 mg PSA (ethylenediamine-N-propylsilane) and 500 mg of MgSO4 gave relatively good recovery rate values for eleven β-blockers, ranging from 71.8 to 121.9%, with relative standard deviations comprised between 2.4 and 12.6%. Similarly, three β-blockers and other pharmaceutical compounds were extracted from fish tissue samples by the QuEChERS approach [73]. After optimization, lower recovery values of β-blockers were obtained compared to previous results, which might be due to the fact that Rodriguez et al. [73] carried out a multi-residue analysis of 24 compounds, and that several parameters were optimized. Three extraction procedures, including PLE, QuEChERS and Ultrasonic Extraction (USE) were tested to extract metoprolol and propranolol from fish samples [29]. After comparing these three extraction methods, it was found that USE was not applicable, since only five of the twenty compounds under study were extracted. However, QuEChERS provided satisfactory recoveries above 40% for most compounds. Finally, PLE was selected as the most appropriate extraction technique, due to the best recoveries obtained.
A solvent extraction procedure of twenty five pharmaceutical products, including three β-blockers, in fish tissues was carried out [74]. Due to the considerable variation of lipophilicity and pKa of pharmaceuticals, a systematic study of extraction behavior was conducted to identify the optimal solvent for the extraction of analytes from fish muscle tissue. Ten solvents, differing by their pH and/or polarity, were tested. The results showed that the extraction efficiency was improved by increasing the organic solvent polarity. In addition, the aqueous-organic mixtures were found to be more efficient, the pH value of the medium having no effect on the β-blocker recovery rates. Thus, a methanol-0.1 M aqueous acetic acid 1:1 mixture (pH 4) was identified as the optimal medium giving recovery values of over 60%.
A new procedure based on the micro-extraction by a packed sorbent (MEPS) for the extraction of three beta- blockers, including metoprolol, labetalol and propranolol, from human urine was developed by Šatínský et al. [75]. The MEPS technique combined sample extraction, purification and enrichment in a single procedure, and was carried out by using a C18 sorbent directly connected in line to an analytical column where the separation was performed. A mixture of acetonitrile - 0.5% triethylamine aqueous solution with acetic acid, the pH value being adjusted at 4.5 in linear gradient elution mode, was used. An on-line MEPS-HPLC coupling showed satisfactory validation results with recovery values ranging from 94.0 to 104.7%, and accuracy in the range of 0.6 to 9.5% for β-blockers. MEPS had significant advantages in terms of speed and simplicity, and it could also reduce matrix effects that often plague the analysis of complex matrices [76]. Recently, a Fast Fabric Phase Sorptive Extraction (FPSE) procedure coated with 20 M CW sol-gel was developed [77]. This was the first study using a Carbowax-modified cellulose tissue extraction membrane that incorporated a sol-gel hybrid inorganic-organic polymer coating for the extraction of six β-blockers in human serum and urine. FPSE was a robust and flexible microextraction procedure with high permeability, providing rapid extraction equilibrium and high chemical stability (pH range: 1-12), without considerable loss of extraction performance. Moreover, several parameters including the extraction time, sample volume, sorbent size, and elution solvent affecting the extraction performances, were systematically investigated. After optimization, mean recoveries for all analytes ranged from 86.4 to 111.3% for all samples with RSDs smaller than 10.7%.
Wu et al. [78] synthesized phosphonic acid functionalized Porous Organic Polymers (PPOPs) used as novel adsorbents for the SPE of β-agonists and β-blockers in milk samples. Before extraction, the milk samples were pretreated with a solution of Trichloroacetic Acid (TCA) in order to finally precipitate the milk-contained proteins. Due to the high adsorption capacity and good selectivity of PPOPs, five β-blockers were identified in milk samples, using the materials prepared for the SPE sample treatment prior to analysis. Thus, the recovery rates of β-blockers in real samples ranged from 62.4 to 119.4 %, depending on the concentration, with relative standard deviations from 0.6 to 12.1 %. The different extraction procedures for β-blockers in various matrices were summarized in (Table 2).
Table 2: Different extraction methods of β-blockers in various samples.
|
β-blocker |
Sample |
Extraction method |
Solvent |
Recovery (%) |
Reference |
|
(R)-(+)-atenolol (S)-(-)-atenolol |
Human plasma |
LLE |
Chloroform/2-propanol (4:1) v/v |
100.2 102.7 |
[79] |
|
Bisoprolol Métoprolol |
Human plasma |
LLE |
Diethyl ether Dichlorométhan |
89 98 |
[53] |
|
Atenolol Bétaxolol Metoprolol Nadolol Propranolol Pindolol Sotalol Timolol |
Bovine eye tissue |
LLE |
Dichlorométhan/ ethyle acétate (1:1) v/v |
89.4 100.3 89.8 83.4 94.1 84.5 53.8 92.7 |
[80] |
|
Alprenolol Labetalol Propranol +4antidepresseurs |
Wastewater |
LLE |
Dichloromethan |
77-113 |
[81] |
|
Carvedilol |
Human plasma |
LLE |
Etherdiethyl and ethyle acetat at pH basic |
85.9-91.1 |
[82] |
|
23 β-bloquants |
Urine |
LLE |
tert-butylmethyl ether |
> 70 |
[83] |
|
Bisoprolol |
Human plasma |
LLE |
Ethyl acetat in NaOH 1M |
86 |
[84] |
|
Atenolol Metoprolol |
Urine |
LLE |
Chloroform |
83 98 |
[85] |
|
Acetobutolol Betaxolol Bisoprolol Métoprolol Nibivolol Sotalol |
Human serum |
LLE |
Butyl acetate |
88 82 96 89 100 89 |
[56] |
|
Pindolol |
Environnement water |
LLE |
Dichloromethane |
99 – 100 |
[46] |
|
(R)-(+)-aténolol (S)-(-)-aténolol |
Human Plasma |
SPE (C18) |
3 ml methanol, 1 mL water |
109 103 |
[79] |
|
Atenolol Bétaxolol Métoprolol Sotalol +liqud regulating agents |
River water |
SPE (C18) |
5 mL methanol, 3mL pure water, pH 10.5 |
48 42 43 63 |
[51] |
|
Bétaxolol Bisoprolol Métoprolol Propranolol Oxprenolol |
River water |
SPE (Oasis HLB) |
6ml methanol, 6ml pure water, pH 7.5 |
102 103 100 94 103 |
[17] |
|
Aténolol Métoprolol Propranolol Sotalolol + other compound |
Surface water |
SPE (Oasis HLB) |
5 mL methanol, 5 mL pure water, neutral pH |
96 103 81 115 |
[59] |
|
Acétobutolol Aténolol Métoprolol Sotalol |
Waste water |
SPE (Oasis HLB) |
2 mL hexane, 2 ml acetone,10ml methanol, pH 10 |
78 101 87 94 |
[63] |
|
(R)-(+)-aténolol (S)-(-)-aténolol E1-métoprolol E2-métoprolol Peak1-nadolol Peak2-nadolol E1-pindolol E2-pindolol (R)-(+)propranolol (S)-(-)propranolol E1-sotalol E2-sotalol |
Wastewater |
SPE (Oasis HLB) |
5 mL methanol, neutral pH |
107 100 85 87 106 56 75 76 86 86 110 81 |
[65] |
|
Aténolol Métoprolol Propranolol Sotalolol |
River water |
SPE (Oasis HLB) |
méthanol and water, pH 2.5-3 |
111 67 54 82 |
[10] |
|
Bisoprolol Métoprolol |
Ground water |
SPE (Oasis HLB) |
3 mL methanol, 3 mL pure water |
114 87 |
[13] |
METHODS OF ANALYSIS OF BETA-BLOCKERS
Liquid Chromatography (LC)
Due to the strong polarity of β-blockers, Liquid Chromatography (LC) was considered as the most suitable method for their analysis [51] because it was easy to use, sensitive, rapid and reproducible [88]. Thus, a number of studies concerned the LC method coupled with MS or in tandem (MS-MS) for the determination of β-blockers in different matrices, such as aquatic environment [7- 90], solid matrices [3-91], biological samples [92-94] and animal food [37-96]. However, some parameters, including the choice of the mobile phase composition, had to be optimized in order to obtain a good separation of the analytes. In most cases, the separation of β-blockers was carried out by HPLC on a C18 column with different mobile phases, consisting mainly of acetonitrile/water and methanol/water mixtures with various buffers.
Finally, to improve the sensitivity of MS detection, buffer solutions, such as ammonium acetate, acetic acid, formic acid or methylammonium acetate, were used [88]. For MS detection, triple quadrupole (QqQ) MS instruments were widely employed for coupling with HPLC in environmental analysis. Indeed, QqQ instruments could help to avoid false positives if ions from at least two ion-ion transitions were used in combination with at least one ion intensity ratio. Moreover, recent developments of hybrid MS instruments achieved high sensitivity, specificity and selectivity since they combined the main advantages of two analyzers,i.e. quadrupole and time-of-flight in the case of QqTOF, and quadrupole and coating ion trap for QqLIT (triple quadrupole-linear ion trap) [97]. Therefore, several articles published in the recent literature have reported the HPLC-QqQ/MS method for multi-class analysis of pharmaceuticals [98,99].
Analysis of Beta-Blockers in Environmental Matrixes
Due to advances in analytical chemistry, the presence of pharmaceutical products in the environment was observed worldwide in recent years. In order to monitor water contamination and to assess the fate of β-blockers, many papers focused on the LC method [12-106]. Generally, the determination of β-blockers in environmental matrixes was carried out by a multi-residue analysis, aimed to simultaneously analyze different families of compounds with various physicochemical properties. This analysis required the optimization of several parameters, such as the nature of the mobile phase, the pH value and mass spectrometry parameters (MRM mode) during the detection of the analytes. In early studies, the analysis of β-blockers in environmental waters was carried out by [19] and [107], who developed a multi-residue LC/ MS/MS method for the detection and quantification of different families of pharmaceutical compounds, including β-blockers. LOQ values of 5 ng/L in surface water and 50 ng/L were found in wastewater for all β-blockers. A comparison of GC/MS and LC-ES/MS/MS methods indicated that only the latter method allowed the analysis of extremely polar β-blockers, such as atenolol and sotalol, because of an incomplete derivatization of functional groups by GC/MS.
A monitoring study on the presence of seventy-four drug residues, including seven β-blockers in natural waters by LC-MS/MS was carried out by Sacher, et al. [108]. Since β-blockers are basic compounds, Electrospray Ionization (ESI) was performed in the positive mode, giving rise to protonated ions at low pH values. After a SPE extraction, the analytes were separated by LC on a C18 column with an ammonium acetate mobile phase (pH 6.8) in an acetonitrile/methanol mixture (2:1, v/v). LOD values in the range 2.4–5.0 ng/L were found. This method was successfully applied to the determination of β-blockers, and their presence was confirmed in German groundwater [108]. Similarly, Hernando et al. [12] utilized this method for the quantification of two classes of pharmaceutical compounds, including β-blockers (atenolol, betaxolol, metoprolol and sotalolol, in WWTP effluents, rivers and drinking water. Due to matrix effects in the wastewater samples, higher LOD values ranging from 17 to 750 ng/L were obtained, with relative standard deviation values, ranging from 3.7 to 18.5%.
Twenty-nine pharmaceutical products of different classes, including four β-blockers, were simultaneously identified by Gros et al. [59] in wastewater and rivers. In this study, the Electrospray Ionization (ESI) source yielded the suppression of the β-blocker mass spectrometry signal, because this signal was very sensitive to other components present in the matrix, leading to signal suppression (or enhancement), and, consequently, erroneous results. Therefore, Gros et al. [59] tested calibration procedures, as well as the dilution of sample extracts. After optimization, it was found that the sample dilution procedure gave the best correction for matrix effects. The method showed a good sensitivity with LOD values between 7.0 and 60 ng/L in wastewater. Other results, obtained by Vieno et al. [63], revealed lower LOD values between 2.1 and 21.0 ng/L, after an external calibration was performed for the correction of matrix effects in wastewater. Also, Gros et al. [61] employed a more sophisticated hybrid mass analyzer, such as QqLIT for the analysis of eight β-blockers in environmental waters by LC-MS. QqLIT analyzers were used successfully to unambiguously confirm the identity of analytes. Very low LOD values between 0.2 and 6.5 ng/L were found in STP effluent and between 0.4 and 6.4 ng/L in STP influent. In addition, the LC-QqLIT/MS method proved to be a powerful analytical tool, since the instrumental detection limits (IDLs) ranged between 0.2 and 2.7 pg, injected in the multiple reaction monitoring mode (MRM). In addition, the inclusion of highly sensitive MS/MS analyzers for each compound, when working in the Information Dependent Acquisition (IDA) mode, provided further confirmation for the unequivocal identification of target compounds in matrixes.
In order to identify the transformation products of β-blockers, resulting from biological and/or physicochemical treatments, [99] developed a LC/MS technique with a quadrupole (Q)TOF-MS analyzer. In this study, the metabolite standards of metoprolol, bisoprolol and propranolol were obtained in vitro by incubation of the parent compounds. The separation of these metabolites in wastewaters was performed by LC after SPE extraction. This method proved to be extremely sensitive, with low very LOD values, comprised between 0.1 and 0.3 ng/L, 0.01 and 0.06 ng/L, and 0.02 and 0.1 ng/L, respectively, for the metabolites of propranolol, bisoprolol and metoprolol.
Because of their similar structures, β-blockers and β-agonists were often analyzed simultaneously in environmental waters. For example, Lee et al. [49] developed a method for the analysis by LC/MS/MS of β-blockers and of β-agonists in treated wastewaters from Canadian treatment plants. A combination of UV and fluorescence detection was initially used to establish the chromatographic separation of compounds. For fluorescence detection, the emission and excitation lengths being set at 310 nm and 230 nm, respectively, because most β-blockers emitted between 300 and 320 nm. UV detection at 220 nm was used for acebutolol, timolol, pindolol and labetalol, which showed a weak fluorescence. The analysts were separated on a Zorbax SB-C 8 column, except for metoprolol and clenbuterol. LOD values ranging from 6 to 11 ng/L were found for these compounds. Similar results were obtained by Salem et al. [7] for the determination of traces of β-blockers and β-agonists in distilled and waste waters by LC-ESI-MS- MS. Drug separation was performed on a C-18 Hypersil Gold column, with a mobile phase, consisting of methanol and ammonium formiate. The β-blockers under study (acebutolol, atenolol, metoprolol, propranolol, timolol, nadolol, labetalol, oxprenolol, pindolol, alprenolol) were identified at retention times ranging from 1.22 to 7.34 min. Drug-spiked wastewaters showed intra-day RSD accuracy values of 3.36-12.49 %, and inter-day RSD accuracy values of 6.42-19.77 %, depending on the concentration. Linear relationships were obtained over the concentration range of 1.0 to 100 ng/L, with LOD values of 0.1-6.7 ng/L for all β-blockers, except oxprenolol.
An ultra-high performance liquid chromatography (UHPLC) improved method was recently developed for the multi-residue analysis of various pharmaceutical compounds, including β-blockers in the environment. UHPLC offered several advantages, such as the use of small diameter particles (typically 1.7 μm) in the stationary phase, and short columns capable of withstanding pressures up to 1000-1300 bar. In addition, this method provided narrow peaks, with a considerable reduction in the analysis time and solvent volume compared to HPLC. Due to the rapid sample preparation and separation of a large number of pharmaceuticals as well as the high sensitivity and selectivity, UHPLC was developed by Gros et al. [109] for the analysis of eighty-one pharmaceutical residues including six β-blockers in various aquatic matrices. UHPLC coupled with quadrupole linear ion trap tandem mass spectrometry (QqLIT) provided LOD values between 0.1 and 11.6 ng/L for β-blockers, except for nadolol and sotalol, with LOD values lower than 0.01 ng/L. These results were in agreement with those of Petrovi? et al. [110] who reported the UHPLC-QqLIT-MS/MS method for the analysis of 81 of various therapeutic groups including six beta-blockers in different aqueous matrices. LOD values were comprised between 0.01 and 0.5 ng/L, 0.1 and 1.5 ng/L and 0.6 and 9 ng/L, respectively in drinking water, surface water and waste water for beta-blockers. Also, the analysis of β-blockers and other compounds was performed in hospital wastewater samples by Gómez et al. [111], using a multi-residue method based on LC/MS/ MS. LOD values were comprised between 28 and 8 ng/L for atenolol and propranolol, respectively. The method was successfully tested on the presence of drug residues, including β-blockers in hospital effluents.
The reuse of wastewater for irrigation and bio-solids (treatedsludge) as fertilizer might leadtothe contamination of agricultural land by pharmaceutical compounds. As a result, research was carried out on their fate in WWTPs and their distribution in different compartments, in order to better understand their behavior in the environment [26-113]. However, few studies were performed on the occurrence of β-blockers in solid matrices. Ramil et al. [3] examined the fate of five β-blockers (pindolol, atenolol, propanolol, sotalol and bisoprolol) in sediments and wastewater by the LC-ESI method in tandem with MS. LOQ values ranging from 1 to 5 ng/g were found by extracting 1g of sediment. The physicochemical properties of β-blockers, such as the adsorption or distribution coefficients, which might influence their fate in solid matter, were investigated. Thus, the sorption of β-blockers was determined experimentally from the dissociation constant Kd values ranging between 0.51 and 4.55 L/kg. It was showed that the β-blockers were weakly absorbed in the solid phase, except for propranolol. In fact, these results were consistent with those obtained by Maurer et al. [112], who reported that propranolol had Kd values between aqueous phase and solid phase (sludge-water) higher by approximately 0.32 L/g solid. Despite their low sorption, beta-blockers might be detected at concentrations up to 86 ng/g (bisoprolol) in sediments. Similarly, Scheurer et al. [91] studied the contamination of WWTP sludge by β-blockers and predicted the adsorbed quantities, using a municipal wastewater treatment plant. Nine β-blockers were analyzed by LC/MS/MS after extraction of solid samples by PLE and by SPE for wastewater. The LOQ values were comprised between 1 and 10 ng/L, and 0.5 to 5 ng/L, respectively, for the WWTP tributaries and effluents.
Finally, in order to elucidate the seasonal concentration levels of cardiovascular drug residues in waste water, a study was carried out by Varga et al. [114]. Nine beta-blockers, including sotalol, atenolol, propranolol, metoprolol and other compounds, were extracted by SPE, followed by LC/MS/MS analysis. The method gave very low LOD and LOQ values, ranging, respectively, from 0.2 to 5 ng/L and from 1.0 to 10 ng/L. Bisoprolol, metoprolol and four other cardiovascular residues were determined in samples taken from four rivers and WWTPs in Serbia by the LC/MS/MS method [13]. The method provided LOD values of 0.33 ng/L for bisoprolol and 1.69 ng/L for metoprolol. A two-phase hollow fiber protected the liquid- phase micro-extraction (HF-LPME) technique, associated with a HPLC-UV procedure was developed for the analysis of six β-blockers in natural water [16]. Under optimal conditions, the extracts were analyzed by HPLC with UV detection. Satisfactory results were obtained, with a good linearity of calibration curves for all β-blockers in the concentration range of 0.1 - 200 ng/mL, and LOD values between 0.08 and 0,5 ng/mL, according to the compound.
A LC/MS/MS analysis coupled with dSPE of six β-blockers in environmental water samples was also performed by [67]. This method was validated over a linear range of 5 to 5000 ng/L, with LOD and LOQ values of, respectively, 17 ng/L and 50 ng/L in tap water. Since the β-blocker enantiomers had different pharmacological activities and modes of action, they presented different therapeutic properties in humans [88]. Moreover, the β-blocker enantiomers of β-blockers were shown to have different ecotoxicological effects [115,116].
Therefore, a chiral analysis in the environment was of great importance to monitor the presence of chiral pharmaceutical residues and the determination of the enantiomeric fraction (EF) which could provide information on toxicological effects [117]. In fact, the separation of chiral β-blockers was previously applied in biological samples and in pharmaceutical formulations by chromatographic and electrophoretic methods [88- 120]. However, LC associated with chiral stationary phases was the reference method because of the variety of commercially available chromatographic columns. For example, [64] developed an HPLC/MS/MS method for the chiral analysis of three β-blockers, including atenolol, metoprolol and propranolol, in the WWTP influents and effluents. The compounds were separated on a chirobiotic V chiral column, with a mobile phase consisting of a 90:10 methanol/water mixture and 0.1% TEAA, adjusted at pH 4.0 with acetic acid. LOD values between 3 and 17 ng/L, and between 17 to 110 ng/L, were obtained, respectively, in effluents and in WWTP tributaries. This method was applied by measuring the stereoisomeric composition of analytes and by determining EF in wastewater. Racemic amounts of the three β-blockers were found in a sewage treatment plant (WWTP), while non-racemic amounts were observed in another station.
An enantioselective multi-residue LC/MS/MS method was developed by [65] for three classes of pharmaceuticals, including β-blockers. Low LOD values, ranging from 0.2 to 7 ng/L were found for the β-blockers in the effluents. A comparative study of different chiral stationary phases (CSPs) was carried out for the analysis of four β-blockers, including atenolol, metoprolol, propranolol and pindolol, by [104]. Four different types of CSPs such as Chiralpak AD-H, Lux Cellulose-1, Chirobiotic T and Sumichiral OA- 4900 were compared, using polar organic (PO) elution mode and normal phase (NP) elution mode. The result showed that Chirobiotic T was the only CSP that resolved all compounds, tested separately, in PO elution mode. The optimized conditions were a mobile phase composed by n-hexane/ethanol/DEA (70/30/0.3, v/v/v) at a flow rate of 1.0 mL min-1 and 25?C. The method was applied to the separation of propranolol enantiomers in natural waters by HPLC-DAD, after extraction by MIP SPE. It was found to be selective, accurate and linear in the range of 0.12-50 μg/mL, with a LOD = 0.4 ng/L for both enantiomers. [121] also studied by HPLC-UV the simultaneous enantiomeric separation using a LuxW Cellulose1/Sepapak-1 as CSP of four β-blockers in natural water samples. The method presented a good linearity (R2 ≥ 0.99), but its sensitivity was relatively low: LOD = 3.0 µg/L for the enantiomers of propranolol and pindolol, and 20.0-22.0 µg/L for the enantiomers of metoprolol and atenolol. It should be suitable for the routine monitoring of emerging pollutants in natural waters. Moreover, [122] developed a LC/MS/MS method to separate the enantiomers of metoprolol and its two metabolites in environmental water. Four different types of chiral stationary phases were investigated for the separation of the eight stereoisomers of metoprolol and its metabolites: Chiralcel OD-H, Chirobiotic V, Chiral AGP and Chiral CBH. In the final method, the enantiomers of metoprolol and four stereoisomers of α-OH-Metoprolol were separated using Chiral CBH, and the enantiomers of COOH-Metoprolol were separated employing Chiral AGP.
The sensitivity of the LC/MS/MS method was determined in treated wastewater samples for these compounds. The method detection limits (MDLs) of the enantiomers metoprolol were 0.96 pM and 2.9 pM for (R) and (S), respectively, and were between 0.23–0.46 pM for the four stereoisomers of α-OH-metoprolol. For the enantiomers of COOH-metoprolol, the sensitivity was lower, MDL was 41.7 pM for both enantiomers. In addition, this is the first time the stereoisomers of α-OH- metoprolol have been detected in wastewater samples with concentrations between 54 and 151 pM for the fourth stereoisomers [122]. Also, a new supercritical fluid chromatography (SFC) method coupled with MS/MS was developed for the chiral separation of atenolol, metoprolol, propranolol and metoprolol acid in environmental matrices [123]. The advantage of SFC relatively to HPLC was that the column efficiency did not decrease with increasing flow rate, contrarily to HPLC. In addition, the SFC utilized less amount of solvent, with a comparable analysis rate. A Chiralpak® IB-3 chiral stationary phase was combined with an Acquity® UPC2 BEH 2-EP column, to avoid interference from metoprolol and its metabolite. The LOD values of the (R) and (S) enantiomers were, respectively, 65.7 and 81.6 ng/L for atenolol, 9.2 and 10.2 ng/L for metoprolol, and 27.5 and 35.0 ng/L for propranolol. The method was successfully applied to monitor the evolution of the enantiomeric fraction with time in a laboratory study on the degradation of wetlands [123]. A novel solid- phase magnetic extraction technique, based on magnetic multi-walled carbon nanotubes, was developed for the simultaneous enantiomeric analysis of five β-blockers in environmental samples by LC-MS/MS [20].
Analytes adsorbed on Mag-MWCNTs were eluted and determined on a chiral α acid glycoprotein column, coupled with triple quadrupole mass spectrometry. LOD and LOQ values of the enantiomers ranged, respectively, from 0.50 to 1.45 ng/L, and from 1.63 to 3.75 ng/L. Another study was performed to determine the concentrations of five β-blocker enantiomers in fifty-eight samples of environmental waters (hospitals, WWTP, agricultural land and canal) [6]. Results showed that the Enantiomeric Fractions (EFs) of metoprolol and propranolol in soil samples showed a trend of enrichment of E1 (first-eluted) compared to E2 (second-eluted), while sotalol was almost racemic. In the drainage canal, no significant difference was found for the pair enantiomers of each β-blocker, while in hospital and WWTP wastewaters, E1 predominated. Recently, the enantiomeric determination of some beta- blockers such as atenolol, metoprolol and propranolol by liquid chromatography tandem mass spectrometry (LC- MS/MS) using a chiral column was reported [124]. The method was optimized and validated for their dosage in soils, composts and digested sludge. Thus, the MDL values in the soil were between 26.5 and 25.8 ng g-1, respectively for the S -(-) and R -(+) of atenolol, between 0.61 and 0.92 8 ng g-1for the enantiomers of metoprolol and between 0.57 and 0.66 ng g-1, respectively for the S-(−) and R-(+) of propranolol.
Analysis of Beta-Blockers in Biological Fluids and Pharmaceutical Formulations
The determination of pharmaceuticals in biological media was essential for therapeutic monitoring and toxicology control. The β-blockers had relatively narrow therapeutic ranges (10 to 1000 ng/ml), and exposure to higher concentrations in this range induced toxicity. In forensic science, β-blockers were routinely found in clinical and autopsy specimens. In some sport competitions, β-blockers were used to minimize heart rate and tremors, in order to improve the performances [125]. For these reasons, β-blockers were considered as doping agents, and they were prohibited since 1988 in sport competitions, such as billiards, golf, cars, etc. by the World Anti-Doping Agency (WADA) in 2021.The detection of β-blockers in biological fluids was essential for anti-doping control, using the CL method for their determination and quantification. For example, [126] analyzed a series of sixteen β-blockers, including atenolol, bisoprolol, metoprolol, propranolol, pindolol, sotalol, in urine samples, by three methods, LC/MS, GC/MS and ELISA. The comparison of the results showed that the ELISA technique could be applied as a screening method, because it provided a good selectivity, but its sensitivity was lower than chromatography. For the chromatographic methods, a good sensitivity, accuracy and precision were obtained, but the GC analysis time was longer, because it required a derivation step. LOQ values between 0.53 and 2.23 ng/mL were obtained with the LC/ MS method.
An anti-doping control study of forty-four compounds prohibited in sport, including twenty-three β-blockers, was carried out by Mazzarino, et al. [83]. Two chromatographic techniques, including hydrophilic interaction liquid chromatography (HILIC) and HPLC, were used for the separation of these compounds. HILIC was mainly used to separate polar compounds, and compounds not enough retained by RP-HPLC. The analytical performances of both methods were compared. The results showed a better sensitivity for the HILIC technique, with LOD values of 50 ng/L for all b-blockers, except aprenolol (80 ng/L). Therefore, HILIC was considered as a powerful approach for the screening and confirmation of β-blockers in sports doping control tests. Santos, et al. [127] also described a LC-MS/MS method for the detection of seven β-blockers (oxopronolol, atenolol, metoprolol, labetalol, propranolol, nadolol and pindolol) in urine for doping control. The analysis consisted to directly inject urine samples into a column of molecularly imprinted polymers, connected to a multidimensional chromatographic system. After optimization of the analytical parameters, the method was found to be linear over the range of 3 to 50 µg/L for all compounds, except oxpronolol (1 to 75 µg/L). The LOD and LOQ values were, respectively, 1 µg/L and 3 µg/L. This method allowed a rapid analysis of these β-blockers, without manual sample preparation, which was essential for routine anti-doping analysis.
More recently, Šatínský, et al. [75] described a novel approach for the automation of sorbent-packed micro-extraction (MEPS) coupled with HPLC. The chromatographic separation was carried out on a C18 analytical column with fluorescence detection (λex = 293 nm and λem = 305 nm). LOD values were 1.4 to 2.8 ng/ mL for β-blockers. Also, six β-blockers were determined in urine by UHPLC/MS/MS, combined with a novel textile phase sorption extraction procedure [77]. The linearity of the method was evaluated in the range of 50 to 5000 ng/ mL with LOD values between 0.3 and 2 ng/mL.
Analysis in Food of Animal Origin (Meat, Kidneys, Liver, Milk, Etc.) β-blockers were administered clandestinely to animals, in order to avoid stress and death, during their transportation to slaughterhouses [41]. In addition, these compounds were injected into animals a few hours before slaughter, which might induce their presence in edible tissues, such as liver, kidneys, muscles and dairy products. To ensure the legal use of β-blockers, the health of consumers and infants, sensitive and selective analytical methods were developed for their detection. Thus, HPLC- MS/MS was developed for the analysis of nineteen β-blockers and eleven sedatives in animal tissues (kidney, liver, porcine and bovine muscles) [96]. A C18 UPLC column was used to separate the analytes, followed by detection in tandem MS, using an electrospray ionization source in the positive mode. The LOQ values were 0.5-2.0 µg/kg for these compounds in various matrices. This method was successfully applied for the screening and confirmation of target drugs in more than two hundred samples. Residues of five β-blockers, including metoprolol, propranolol, betaxolol, labetalol and penbutolol, and twenty-three β2-agonists were simultaneously quantified in animal muscle tissues by the HPLC method [66]. Methanol and 0.1% formic acid were used as mobile phases for gradient elution. A Waters Atlantis®T3 column was used for the separation. The ESI positive ion scan mode was used with selective reaction monitoring. The linearity of the method for β-blockers went from 5 to 200 µg/L, with correlation coefficient (r2) values around 0.995, and the LOD values of β-blockers for muscle tissues were 0.1 - 0.2 µg/kg. This method was sensitive and specific for the determination of β-blockers in muscle samples from pigs and chickens.
Sai et al. [95] reported a LC/MS method for the analysis of twenty-three β-blockers and twenty-five β2-agonists in pork meat samples. The method was linear over the concentration range of 5 to 200 µg/L ( r2 > 0.99). For the forty-eight analytes under study, the LOD values in muscle, liver and kidney samples ranged from 0.05 to 0.49 µg/kg, and the LOQ values were comprised between 0.13 and 1.64 µg/kg. This method was successfully applied to one hundred and ten real-source food samples, including pork and chicken meat, liver and kidney samples. Similarly, residues of nine β-blockers, including atenolol, pindolol, acebutolol, metoprolol, carazolol, labetalol, bisoprolol, propranolol and penbutolol, were extracted from dairy products (fluid milk, powder, cheese) by d-SPE followed by a LC separation with MS/MS detection [128]. It was validated in the range of 0.1 - 20.0 µg/L, with LOD values of 0.5 µg/L. Recovery rates for samples spiked at three levels (0.5, 1.0, and 5.0 µg/kg) ranged from 89.5% to 112.5%, with RSD values between 4.2% and 11.5%. Also, Shang et al. [72] developed a new method for the determination of eleven β-blockers in meat by QuEchERS extraction coupled with UPLC-MS/MS. The separation was carried out on a C18 column with detection by electrospray ionization in the positive ion mode. The linear concentration range was 1 to 500 μg/L (r2 > 0.999). The LOD and LOQ values ranged, respectively, between 0.02–0.16 μg/kg and 0.07–0.54 μg/ kg. This method was applied to the analysis of β-blockers in twenty-two real samples. No target β-blockers were detected in those samples. The results showed that the use of illegal β-blockers occured rarely in sedative functional foods.
Similarly, Zhang et al. [129] analyzed two β-blockers (propranolol and penbutolol) and fourteen β-agonists in porcine muscle. Under optimal conditions, the LOD values were 0.94 μg/kg for propranolol and 0.38 μg/kg for pendotolol. Moreover, de Oliveira et al. [68] developed a LC-MS/MS method for the analysis of fourteen β-blocker residues in porcine, bovine and equine kidney. All samples were extracted with acetonitrile, and purified by dispersive solid phase extraction (d-SPE). Similarly to previous studies, the LOD values were 1.25 μg/kg for atenolol, carvedilol, nadolol and penbutolol, 0.25 μg kg-1 for carazolol, and 0.5 μg kg-1 for other β-blockers. The LOQ values were 1.25 μg/kg for carazolol and 2.5 μg/kg for the other compounds.
The presence of twenty-six β-blockers, including six metabolites, in infant milk powder was demonstrated by a new HPLC-Q-Orbitrap HRMS method [40], which gave a linear relationship over the range of 0.5 to 500 μg/kg, with LOD and LOQ values ranging, respectively, from 0.2 to 1.5 µg/kg and from 0.5 to 5.0 µg/kg. Also, Wu et al. [78] developed a simple and sensitive method for the detection of five β-blockers and ten β-agonists in milk samples by ultra-high performance liquid chromatography coupled with high-resolution tandem mass spectrometry (UPLC-HRMS), associated with SPE phosphonic acid- functionalized porous organic polymers (PPPO). A RP- MS Accucore reverse phase column was used for analyte separation. The mobile phase consisted of water + 0.1% formic acid and methanol. It showed a good linearity within 3-5 orders of magnitude (r2 = 0.995 - 0.999). The LOQ values ranged from 0.05 to 0.25 ng g-1, and the LOD values from 0.02 to 0.1 ng g-1. Therefore, this method could be a valuable tool in regulatory laboratories for the control of illegal use of these drugs in dairy products.
Analysis in Biotic Species
Presently, only few chromatographic studies were performed on the detection and bioaccumulation of β-blockers in some aquatic species, such as fish, bivalves and mullet. The accumulation of pharmaceutical products in biological tissues could be partly linked to non-ionized species, with a strong affinity for lipophilic materials remaining in the aqueous phase. Therefore, benthic species, such as marine bivalves, might bio-accumulate pollutants present in the dissolved phase (through the gills) or adsorbed on the colloids (through the digestive tract). LC methods were generally employed for the detection of β-blockers in biotic species. For example, Ramirez, et al. [74] developed a screening LC-MS/MS method targeting twenty-three pharmaceutical products and two metabolites, including three β-blockers (atenolol, metoprolol and propranolol) in fish tissues. The reverse phase separation of target compounds was performed using a C18 column and a nonlinear gradient [0.1% (v/v) formic acid + methanol]. Eluted analytes were introduced into the mass analyzer using positive electrospray ionization for β-blockers. After optimization, the method was linear over the range of 1.25 to 160 ng g-1 for atenolol and metoprolol, and of 0.625 to 60 ng g-1 for propranolol. LOD values were less than 3 ng g-1 for the three β-blockers. It was also used to screen the target analytes in fish from a stream.
A new multi-residue extraction method, based on pressurized LPE and SPE, followed by UHPLC analysis and coupled with triple quadrupole MS, was developed by Wille et al. [87] for the bioaccumulation of fourteen pesticides, ten perfluorinated compounds (PFCs) and eleven pharmaceuticals in marine blue mussel tissues. Atenolol and propranolol were separated on a C18 column with a mobile phase including 0.08% aqueous formic acid, 0.08% formic acid in acetonitrile and isopropanol. Very low LOD values (1 ng g-1) were found for both β-blockers in spiked samples. Huerta et al. [29] reported a sensitive UHPLC– MS/MS method, combined with LPE, for the determination of twenty pharmaceutical compounds and metabolites, including five β-blockers, in fish homogenate, liver and muscle of mediterranean rivers. A thorough evaluation of matrix effects was carried out, in order to select the best approach for the analysis of complex matrices. LOD values of β-blockers were in the ranges: 0.03-0.50 ng/g for fish homogenate, 0.03-0.18 ng/g for fish muscle and 0.09-0.77 ng/g for fish liver.
Moreno-González et al. [28] used the same procedure for the analysis of twenty pharmaceutical products, including six β-blockers, in fish (muscle and liver) and marine mollusks (bivalves and sea snail). After LPE extraction, the separation of the β-blockers was performed on an AcquityHSST3 column with a mobile phase made of methanol + 10 mM formic acid/ammonium formiate (pH 3.2). LOD values were obtained between 0.1 and 0.2 ng g-1 for fish muscle and between 0.1 and 1.9 ng g-1 for fish liver. More recently, twenty-four pharmaceutical products, including β-blockers, were analyzed in various species of mollusks by the LC/MS/MS method [73]. Linear relationships, obtained by the standard addition method, were found over the ranges: 0.58-18.5 ng g-1 for atenolol and 0.68-21.8 ng g-1 for propranolol, with LOD values below 1 ng g-1 for all β-blockers.
Gas Chromatography (GC)
GC was the first method developed for the analysis of β-blockers. However, since these compounds were very polar, their derivatization was a prerequisitefor GC analysis, which was less recently utilized for the determination of β-blockers. Nevertheless, several derivatization procedures, combined with GC, were reported for block- active protons in hydroxy, amino and other polar groups, leading to volatile- and thermally-stable derivatives [125], and to cyclized silylation, acylation and benzoylation [130]. Also, Huggett et al. [102] proposed a GC/MS method for the propranolol, metoprolol and atenolol analysis in wastewater. LOD values were 1 ng/L for propranolol and metoprolol, and 10 ng/L for nadolol.
Five β-blockers, including oxprenolol, metoprolol, propranolol, bisoprolol, and betaxolol, were analyzed in effluents wastewater treatment plants (WWTP) by GC coupled with mass detection (GC–MS) [17]. After extraction of by ESP, a derivatization step, such as trimethyl- silylation with MSTFA (N-methyl-N-(trimethylsilyl)- trifluoroacetamide), followed by a trifluoro-acetylation with MBTFA (N-methyl-N-(trifluoroacetamide), was carried out. Linear relationships were obtained over the concentration range of 1- 400 µg L-1, and LOD values were about 3 ng L-1 for river waters. Due to matrix effects, LOD values were higher in the effluent samples (about 15 ng L-1).
Caban et al. [131] established a comparative study of derivatization procedures for the GC-MS determination of six β-blockers (acebutololol, atenolol, metoprolol, nadolol, propranolol, pindolol) and two β-agonists. Nineteen different derivatization reagentswereusedto obtain asingle derivative for each compound. In addition, the influence of reaction time and temperature on the efficiency of the chemical conversion of compounds were investigated. Five derivatization procedures, including trimethyl- silylation, tert-butyl-dimethyl-silylation, acylation, sequential derivatization and cyclized silylation, were tested. It was showed that the application of the BSTFA+1% TMCS mixture was the most effective derivatization procedure. Moreover, Caban et al. [132] proposed a new silylating agent, DIMETRIS (i.e dimethyl (3,3,3-trifluoropropyl) silyl-diethyl-amine), for the derivatization of β-blockers and β-agonists.
This agent had strong nucleophilic properties, and reacted selectively with hydroxyl groups. Thus β-blockers were determined in tap water by SPE-GC-MS (SIM), with LOD values between 3 and 40 ng L-1, which confirmed that DIMETRIS was suitable for the analysis of pharmaceutical product traces in natural samples and constituted an interesting alternative to silylating and acylating agents. Later, Caban et al. [62] developed a multi-residue method for the simultaneous determination of seventeen human pharmaceuticals, including six β-blockers in drinking waters by SPE-GC-MS (SIM) method with DIMETRIS. The LOD and LOQ values ranged, respectively, between 0.6 and 5.7 ng L-, and between 1.5 and 17.1 ng L-1 for all β-blockers.
Two derivation techniques were also tested for the GC analysis of β-blockers in urine. The first one consisted of silylation reactions of β-blockers, leading to the formation of trimethylsilyl (TMS) and trifluoro-acyl (TFA) derivatives, while the second one was performed with Methyl-Boric Acid (MBA). The LOQ values strongly depended on the nature of the β-blocker, ranging from 0.30 ng/mL for sotalol to 102 ng/mL for labetolol, and from 6.0 ng/mL for alprenolol to 39.6 ng/mL for acebutolol.
An analytical procedure was developed and validated for the quantification of carvedilol in human plasma [82]. Carvedilol and atenolol (internal standard) were extracted from human plasma by LLE with a mixture of diethyl-ether and ethyl acetate at basic pH. Extracts were derivatized with n-methyl-n-(trimethylsilyl) trifluoroacetamide, and analyzed by GC-MS. The calibration curves were linear over the concentration range of 15 -500 ng/mL. The intra- day and inter-day precision, expressed as RSD, was less than 8.0%, and the relative error was about 11.0%. The LOD and LOQ values for carvedilol were, respectively, 5.0 and 15 ng/mL.
GC methods were also reported in the enantiometric analysis of chiral β-blockers, using chiral derivatization reagents. Enantioselective GC methods had advantages, such as simplicity, speed and use of less solvent amount. For example, an enantioselective method was developed for the separation of two propranolol enantiomers in wastewater after GC derivatization, without using an enantioselective stationary phase [133]. Both propranolol enantiomers were derivatized with a chiral compound single enantiomer of (-)-(α)-methoxy-α- (trifluoromethyl) phenyl-acetyl-chloride ((-)-MTPA-Cl) to form diastereoisomers, which had different physical properties, and could be separated on a reversed-phase GC column. Propranolol LOD values were comprised between 0.1 and 1.0 ng/L, depending on the sample. A new micellar electrokinetic chromatographic (MEKC) method was also proposed and validated for the analysis of carvedilol and propranolol in human urine samples [134].
In this study, a vortex-assisted liquid-liquid extraction (VALLE), coupled with amplified field sample injection and sweeping, was used for biological sample cleanup and MEKC sensitivity enhancement. This method was linear over a dynamic range of 0.005 to 1 µg/mL in urine. The intra- and inter-day relative RSD and relative error values of the method were below 20%, indicating a relatively good precision and accuracy. Finally, the method was successfully applied to the analysis of real urine samples.
Spectrophotometric and Spectrofluorimetric Methods
UV-VIS Spectrophotometry: UV-VIS spectrophotometry was a simple, fast and inexpensive method, frequently used for the qualitative and quantitative determination of β-blockers, and their routine analysis. This method was simple, fast and inexpensive, and could be very useful for routine analysis of bulk samples and formulations. Al-Ghannam [135] developed an indirect spectrophotometric method for the determination of atenolol, nadolol and timolol, based on the interaction of drugs with sulfo-phthalein acid dyes, such as Bromophenol Blue (BPB), Bromothymol Blue (BTB) and Bromocresol Purple (BCP), to form yellow, stable ion pair complexes. Under optimal conditions, the three β-blockers could be analyzed in the concentration range of 1 to 10 µg/ml. The lowest LOD values were obtained with BTB dye for nadolol and timolol (respectively, 0.038 µg/mL and 0.085 µg/mL) and, with BSP dye, for atenolol (0.1165 µg/mL).
Also, Gölcü, [136] reported a direct spectrophotometric method for the estimation of acebutolol, atenolol and propranolol in pharmaceutical formulations. No extraction, derivatization or evaporation steps, and no harmful chemicals were necessary for this method, which reduced the analysis time. This method was linear over a concentration range of 37.3 to 111.9, 53.3 to 213.1 and 14.8 to 51.8 µg/mL, respectively, for acebutolol, atenolol and propranolol. The LOD values varied between 0.77 to 1.6 µg/ mL, according to the β-blocker. More recently, Vaikosen et al. [137] proposed an indirect spectrophotometric method for the analysis of atenolol and propranolol in pharmaceutical formulations. Both β-blockers were oxidized with a KMnO4 (2x10-3 M) alkaline solution. After optimization, low LOD values of 0.50 and 0.58 μg/mL were, respectively, found for atenolol and propranolol.
An indirect spectrophotometric method was also used for the determination of atenolol, as pure compound and as pharmaceutical formulation, by Zakaria et al. [138]. The method was based on the oxidation of atenolol by an excess amount of N-bromosuccinimide (NBS). Afterwards, the excess of NBS served to bleach the color of methyl red dye (MRD), and the absorbance of remaining MRD at a wavelength of 518 nm was measured. The absorbance of the unbleached color of MRD corresponded to the atenolol concentration in the sample solution. The Beer’s law was observed in the range of 0.1-2.0 μg/mL, with a molar absorptivity value of 8.89 x 104 L. mol-1. cm-1. This method was applied to the assay of atenolol with satisfactory results.
Spectrofluorimetry: Because of its low sensitivity, spectrophotometry was very limited for the analysis of complex samples, containing them at trace level. Moreover, the lack of specificity of UV-VIS absorption spectrophotometry generally prevented the application of this method, because of spectral overlap. Therefore, spectrofluorimetry was one of the analytical methods that could improve the sensitivity and selectivity of drug analysis. It was often used for the analysis of drugs in biological and environmental media because of its selectivity, its high sensitivity, accompanied by a good precision.
In addition, spectrofluorimetry was less expensive and faster than most other analytical methods. Since a number of β-blockers exhibited natural fluorescence, several studies described their analysis by direct fluorescence measurements, derivative reactions, fluorescence quenching or synchronous fluorescence in biological fluids and pharmaceutical formulations. Also, spectrofluorimetry was often associated with chemometrics, which simultaneously took into account several analytical parameters.
Xu et al. [139] reported a simple, rapid and highly sensitive spectrofluorimetric method for the determination of carvedilol. The relative Fluorescence Intensity (IF) measurement was performed using excitation/emission wavelengths of 254 nm/ 356 nm. The effects of organic solvent, pH and foreign ions on the quantitative analysis of carvedilol were examined. A linear relationship was found between IF and carvedilol concentration in the range of 0.50-270 ng/mL (r2 = 0.999 and RSD = 2.3 %). The LOD was 0.19 ng/mL with recovery rates between 98.7 and 102.1%. This method could be used for the determination of carvedilol in commercial tablets. Also, a higher LOD of 1.7 ng/ml was obtained by combining fluorescence spectrometry with an ionic liquid (IL) micro-extraction procedure for the quantitative analysis of carvedilol traces in pharmaceutical and biological media [140].
In addition, a Flow Injection (FIA) spectrofluorimetric method was reported for the analysis of carvedilol in pharmaceutical formulations [141]. These studies were performed in micellar media by testing several types of surfactants. The effect of the sample injection rate of the FIA system was investigated along with other parameters. In these conditions, carvedilol exhibited excitation/ emission wavelengths of 286/341 nm. The carvedilol IF was enhanced in a SDS medium. The method was linear in the range of 9x10-8 -1x10-6 mol/L with a LOD of 3.63x10-9 mol/L. In addition, this method showed a great selectivity with respect to excipients commonly used in pharmaceutical products. Later, Silva, et al. [142] used Single-Walled Carbon Nanotubes (SWNTs) for the separation of carvedilol stereoisomers by a FIA spectrofluorimetric method. A good separation of the enantiomers carvedilol R (+) and S (-) was obtained with a resolution factor of 3.16. The enantiomer recovery rates were 101.5 and 97.8 %, for S and R, respectively. This method was selective, sensitive, simple and inexpensive for the determination of carvedilol enantiomers and did not required a tedious and time-consuming derivation step.
Also, another spectrofluorimetric method combined with chemometrics was developed for the simultaneous determination of propranolol and levodopa in urine by Madrakian, et al. [47]. The natural fluorescence of these drugs was investigated in micellar media, such as SDS. Several parameters, including the effects of pH and SDS concentration on fluorescence, were optimized. The propranolol concentration increased slightly with pH from 1 to 6 and, then, decreased in an alkaline medium. In SDS micellar media, the propranolol IF value was enhanced 2.4 times, compared to the aqueous medium. Thus, under optimal conditions (pH 4 and [SDS] = 0.02 mol/L), a recovery rate of 99.6 % was found. Good linearity between 3.6.10--9 and 1.8 10-6 mol/L was obtained with an LOD value of 0.20 µg/L for propranolol.
Native fluorescence of Sotalol Hydrochloride (SOT) was measured in tablets and spiked human plasma at excitation/emission wavelengths of 235/310 nm. The LOD and LOQ values were, respectively, 0.37 and 1.08 ng/mL This method offered a high sensitivity, allowing the determination of SOT in human plasma, even in very small amounts, [143]. Recently, a spectrofluorimetric method was developed by our team for the quantitative analysis of pindolol in environmental waters [46]. In this study several parameters such as the effect of solvent, pH and cyclodextrins were studied. Pindolol exhibited natural fluorescence at excitation/emission wavelengths of 260/303 nm. A good improvement in the pindolol fluorescence signal was observed in cyclodextrin medium compared to aqueous medium, with an enhancement factor of ten times in HP-β-CD. After optimization of the analytical parameters, the results showed that the solvent 2-propranol offers the best analysis conditions with very low LOD and LOQ values of, respectively, 0.21 and 0.72 ng/mL. For the validation of spectrofluorimetry, the analysis of pindolol was carried out in spiked natural water samples, using the standard addition method. Thus, spectrofluorimetry might be considered as an alternative method for the analysis of β-blocker traces, in environmental waters.
Three β-blockers, including arotinolol, atenolol and labetalol, were analyzed in human plasma by spectrofluorimetry [144]. The method was based on the reaction of these drugs as electron donors with the fluorogenic reagent 9,10-Dimethoxy-2-Anthracene Sulfonate (DMAS) as a π-acceptor in acid medium. The obtained ion pairs were extracted in chloroform, and were measured at excitation/ emission wavelengths of 385/452 nm. A linear calibration curve was obtained on the range values of of 0.5-5, 1.0-11.0 and 0.6-6.4 µg/mL for labetalol, atenolol, and arotinolol, respectively. This method was successfully applied to the analysis of tablets and to in vitro drug assay in enriched human plasma with recoveries (n = 3) ranging from 96.98 ± 1.55 to 98.28 ± 2.19%.
A new extraction method combined with spectrofluorimetry (SPE) was developed for the determination of atenolol in human urine [44]. After SPE extraction, atenolol was analyzed by spectrofluorimetery (λ ex = 277 nm /λem = 300 nm). The calibration curve was linear between 150 and 4000 ng/m, and the LOD and LOQ values were, respectively, 30 and 100 ng/mL. The SPE method was also applied to the determination of atenolol traces in pharmaceutical and biological samples. Similarly, [45] developed this method for the extraction of atenolol in urine by imprinted polymers, used as SPE sorbents. The calibration curve was linear over the range of 100 to 2000 ng/mL, with LOD and LOQ values close to those obtained by [44]. This method was selective, sensitive and simple for the direct determination of atenolol in human urine. The atenolol fluorimetric determination was also performed by using gold nanoparticles (AuNPs) [145]. This method was based on the atenolol quenching effect on the AuNPs photoluminescence at λem = 705 nm. Parameters affecting the gold nanoparticles liminescence, such as solvent, pH and surfactant, were optimized. A linear range was obtained between 1000 and 10000 ng/mL, with LOD and LOQ values of, respectively, 870 and 2640 ng/mL.
A fluorescence quenching method was also described for the sotalol analysis in urine, plasma samples and pharmaceutical formulations by [146]. The fluorescence quenching of Cucurbit [n]uril (CB[n], n = 7] and palmatine complex by sotalol was measured. There was a competition between sotalol and palmatine molecules to occupy the CB[7] hydrophobic cavities. In these conditions, sotalol occupied the CB[7] cavity and forced palmatine molecules to reside in the aqueous environment. The method, which was precise, simple, fast and reproducible, provided a linear between 100 and 30600 ng/mL with a LOD of 330 ng/mL.
Zhang, et al. [147] determined propranolol in biological samples by fluorescence. The method was based on the fluorescence inhibition of 3-mercaptopropionic acid CdTe quantum dots by propranolol, which was attributed to the synergistic action of the Internal Filter Effect (IFE) and the dynamic quenching effect. Moreover, some common excipients, propranolol analogs, and stimulants did not have a significant effect on the fluorescence signal of CdTe- QDs (Cadimium Telluride Quantum Dots). Overall, the fluorescence sensor had a good stability, a rapid detection ability, a high sensitivity and selectivity, and a wide concentration linear range (0.1 and 200 µM). The LOD value was 0.055 µM. Therefore, this fluorescence sensor might be successfully used for the detection of propranolol in biological samples.
Synchronous fluorescence was often used for the simultaneous analysis of β-blockers, alone or with other compounds. For example, a simple, rapid and accurate synchronous fluorimetric method was developed for the simultaneous analysis of propranolol and pindolol in urine and in pharmaceutical formulations [148]. Propranolol spectra exhibited two excitation wavelength maximums at 232 and 295 nm and two emission wavelength maximums at 324 and 337 nm, whereas pindolol showed two excitation wavelength maximums at 223 and 281 nm and a maximum fluorescence at 308 nm. Since the propranolol and pindolol fluorescence spectra overlapped, synchronous zero-first-derivative spectrofluorimetry was used for the simultaneous determination of these compounds with Δλ = 18 nm. The method was linear over the ranges of 20-1000 ng/mL and of 40 -1200 ng/mL, respectively, for propranolol and pindolol. The LOD values were 3 ng/mL for propranolol and 7 ng/mL for pindolol. Mean analytical recoveries in urine samples were 96 ± 3 and 97 ± 3%, respectively, for both compounds. Similarly, carvedilol and sodium ampicillin were assayed simultaneously by synchronous fluorescence [149]. The fluorescence spectra of carvedilol and ampicillin exhibited one excitation wavelength maximum at 254 nm and two peak emission wavelengths at 357 and 426 nm, respectively, and partially overlapped. Therefore, synchronous fluorescence was used with a Δλ = 80 nm to simultaneously determine both drugs without a separation procedure. Satisfactory recovery rates of 96-103% and linear relationships between 5 and 100 ng/mL were obtained, with a LOD value of 1 ng/mL for carvedilol.
Walash et al. [150] reported a synchronous fluorescence method for the simultaneous analysis of binary mixtures of metoprolol and felodipine. The synchronous fluorescence intensity of both drugs was measured at Δλ = 70 nm in aqueous solution, and different experimental parameters on the synchronous fluorescence intensity of both drugs were carefully studied and optimized. Fluorescence intensity curves were linear over the range of 500-10000 ng/mL for metoprolol. The LOD and LOQ values were, respectively, 110 ng/mL and 320 ng/mL. Also, Mabrouk et al. [151] used synchronous fluorescence for the separation of metroprolol and amlodipine in pharmaceutical formulations. An optimal Δλ value of 90 nm provided a good resolution for both drugs. The method was linear over the range 0.5–10 μg/mL for metoprolol, with a LOD value of 130 ng/mL.
Also, the β-blocker atenolol and the anti-inflammatory drug, diclofenac, were determined simultaneously by first derivative synchronous fluorescence in drinking water samples [152]. The optimal Δλ values were, respectively, 70 and 80 nm for atenolol and for diclofenac. A linear relationship between 4 and 3000 ng/mL was found for atenolol with a LOD value of 1.319 ng/mL. The derived synchronous spectrofluorimetric method was successfully applied to the simultaneous determination of atenolol and diclofenac in drinking tap water samples without prior separation.
Recently, a novel, facile, rapid, and precise synchronous fluorescence method was described for the simultaneous determination of bisoprolol fumarate and ivabradine in dosage forms and in biological fluids by Abo Elkheir et al. [153].The first derivative synchronous fluorescence spectra of ivabradine and bisoprolol fumarate in ethanol were characterized by Δλ = 80 nm. The method was optimized in different solvent systems, pH values, and surfactants, and it was linear for concentration ranges of 30–200 ng/mL and 30 –180 ng/mL, for ivabradine and bisoprolol fumarate, respectively, with LOD values of 4.88 and 5.28 ng/mL.
DISTRIBUTION OF BETA-BLOCKERS
In WWTPs, Surface, Underground and Drinking Waters, Sludge and Sediments
In recent years, emerging contaminants, such as pharmaceuticals, received a great attention in environmental and toxicological research. Indeed, several classes of pharmaceutical products, including β-blockers, were found in different environmental compartments. Due to the high prevalence of cardiovascular diseases, β-blockers were commonly pre-registered for the treatment of hypertension, cardiac arrhythmia, etc. After their ingestion by patients, β-blockers were generally eliminated under their initial form and metabolized in urine. Pharmacokinetic data showed that human excretion rates of these unmetabolized “parent compound” drugs vary greatly depending on the compound. For example, propranolol have the lowest excretion rate in its initial form around 1% [49], whereas 60-70% of pindolol was metabolized in urine. In contrast, 80-90% of sotalol was eliminated under its initial form in urine, which indicated that this drug should come out almost intact of the human body.
Therefore, these drugs ended up in domestic and hospital wastewaters after their excretion by patients, and they were transferred via the sewer in WWTPs, where they underwent different types of treatment. However, WWTPs were found to be ineffective for the total elimination of these micro-pollutants. Various studies showed that pharmaceuticals, such as β-blockers, were strongly hydro-soluble, persistent and poorly degradable. Therefore, they were continuously discharged into surface waters. In addition, during treatment in WWTPs, these compounds could be partially adsorbed on residual sludge, and contaminated the soil after spreading on agricultural lands. Moreover, in developing countries where there was no sanitation system, wastewaters were even directly discharged into the environment. Consequently, the incomplete treatment of wastewater, as well as its direct discharge, constituted the main sources of diffuse pharmaceutical products in the environment.
Thanks to the development of high-performance analytical techniques, pharmaceutical residue traces were detected and quantified in different environmental compartments. Indeed, β-blockers were detected worldwide in influents and effluents of WWTPs, surface water, groundwater and solid matters (sediments and soils). Moreover, β-blockers ranked third among the most commonly pharmaceuticals present in the environment, with concentration levels in the ng/L and µg/L ranges [154]. The β-blocker concentrations in environmental compartments around the world were quite different, because of differences in social and environmental factors, and in processing methods and operating parameters, depending on the β-blocker type and amount. For example, in the USA, β-blockers were in the top two hundred most prescribed drugs, and, consequently, they were often detected in environmental waters [4]. In Germany, it was estimated that more than one hundred tons of these compounds were consumed per year [3]. Yi et al. [22] listed more than twelve β-blockers present in the environment around the world, the most frequently detected being atenolol, bisoprolol, metoprolol, propranolol, and sotalol.
In Europe, Ternes et al. [19] performed the first studies on the contamination of German rivers and sewage by β-blocker residues. For instance, [107] detected four β-blockers in Germany WWTP effluents at concentrations ranging between 0.2 to 2.2 µg/L. Also, [155] studied the fate of seven β-blockers submitted to conventional WWTP treatment in Germany, by measuring their concentrations in influents and effluents. Sotalol and metoprolol were the predominant species, with maximum concentration levels of 1.2-1.3 µg/L in the influents and 1.1-1.2 µg/L in the effluents, which indicated their relatively low degradation rate in WWTPs. Higher concentrations were found by Nödler et al. [156], who quantified three β-blockers, including atenolol, metoprolol and sotalol, and two transformation products of atenolol and metoprolol in surface water, groundwater and wastewater from treatment plants in Germany. Metoprolol and sotalol were detected at concentrations of 6.0 and 4.3 µg/L, respectively, in WWTPs. The atenolol transformation product, i.e., atenolol acid, was detected at a concentration of 12 µg/L, whereas the atenolol concentration was 0.6 µg/L in wastewater. Only atenolol was present in groundwater. Similar results were obtained for six β-blockers in WWTPs, with high metoprolol concentrations of 4.72 µg/L and 4.47 µg/L, respectively, in the influents and effluents [157]. β-blockers were also detected in solid matter, such as scraps and sediments [3], and their physicochemical properties, like adsorption and distribution coefficients, which could influence their fate in solid matter, were investigated. The sorption of β-blockers was experimentally determined from the dissociation constant Kd, with values of 0.51 L/g for pindolol and 4.55 L/g for propranolol.
β-blockers, such as metoprolol, propranolol and bisoprolol, were also quantified in the French WWTP effluents, with maximum concentrations of 1.7, 1.1 and 2.8 µg/L, respectively [17]. [158] also showed the presence of ten β-blockers and evaluated the efficiency of WWTPs. β-blockers were found in all wastewater samples at highest concentrations of 9.8 and 8.4 µg/L, for acebutolol and atenolol, respectively. Later, Gabet-Giraud et al. [2] investigated the presence of ten β-blockers in eight WWTP effluents and in thirty-four surface water samples. The β-blocker concentrations in WWTP effluents were much higher than those determined in rivers, which might be due to the dilution of WWTP effluents in the receiving waters, whereas lower concentrations were also noted in groundwater. The presence of ten β-blockers in effluents and river waters in France was reported by Jacquet et al. [159]. Atenolol was found at maximum concentrations of 2 µg/L in effluents and 0.6 µg/L in river water. Drinking water was weakly contaminated by β-blockers, such as atenolol found at very low levels at 2 ng/L [160].
In Italy, Verlicchi et al. [161] quantified atenolol and sotalol at maximum concentrations of 5.8 μg/L and 5.1 μg/L, respectively, in hospital effluents. Morever, much lower concentrations of 0.008 µg/L for timolol and 0.47 µg/L for atenolol were measured in WWTP effluents, a study on the seasonal variation of β-blockers in water indicating that their concentrations were higher during winter than summer [162]. Recently, (Papagiannaki et al. [163] reported the presence of 0.48 µg/L of atenolol in raw and treated drinking water sources in the metropolitan area of Torino.
β-blockers were also detected in Spain, in various environmental matrices. For example, (Gros et al. [61] reported the presence of five β-blockers in wastewater, with atenolol highest concentrations of 2688 µg/L and 1370 µg/L, in influents and effluents, respectively,. Later, Gros et al. [109] determined atenolol and nadolol in seawater at much lower concentrations of 6.2 and 0.3 ng/L, respectively. Nadolol was detected in drinking water at a concentration of 0.4 ng/L. [164] quantified atenolol, sotalol, metoprolol and propranolol at maximum concentrations of 0.06 to 1.82 µg/L in river waters. Similarly [165] reported the presence of propranolol in surface waters of the Donana National Park (Spain), and in WWTP effluents around this park. Propanolol maximum concentrations of 0.51 and 0.14 µg/L were found in effluents and surface water, respectively, which showed that propranolol underwent a weak photodegradation.
Propranolol concentration was also measured at approximately 0.39 µg/L in the effluents of a municipal wastewater treatment plan [166]. Moreover, López- Serna et al. [70] reported the presence of six β-blockers in Spanish river waters at concentrations between 1.3 and 475 ng/L. García-Galán et al. [167] showed the presence of metoprolol and its metabolites, and transformation products due to wastewater treatment. The ratio (MTPA/ MTP) of metoprolol (MTP) and its metabolite, metoprolol acid (MTPA) varied between 60 and 80 % in the influents, in agreement with the metabolic excretion rates of these compounds (60-65% of MTP excreted as MTPA). The MTP transformation product (α-OH-MTP) presented a higher concentration than that of MTP in untreated wastewater, and in the effluents, the MTP concentration was much higher. Vazquez-Roig et al. [168] reported the presence of atenolol enantiomers in WWTPs in Valencia. A highest concentration of 1322 ng/L of R (+)-atenolol was found in the sewage treatment plant. Moreover, the chiral fate of atenolol was studied during its treatment by means of the enantiomeric fraction (EF). Atenolol was found to be enriched in S (-)-atenolol in raw wastewater, and in the R(+) or S(-) enantiomer during wastewater treatment, depending on the WWTP technology used.
In Greece, Papageorgiou et al. [169] studied the seasonal presence of atenolol, metoprolol and propranolol in environmental waters. Atenolol was the most prevalent compound with peak concentrations of 2346 ng/L and 1707 ng/L, respectively, in the influent and effluent. Recently, Ofrydopoulou et al., [170] quantified three β-blockers (atenolol, bisoprolol and metoprolol) in WWTPs. Atenolol and metoprolol had the highest concentration of, respectively, 815.6 and 280 ng/L in effluents.
The highest atenolol concentrations in Europe were detected in the South Wales (UK), with levels between 33.1 and 7.6 μg/L, respectively, in the influents and effluents of the sewage treatment plant Cilfynydd [170]. In addition, Leusch et al. [171] performed a quantitative comparative study to quantify several micropollutants, including pharmaceutical compounds, in treated wastewater, surface water and drinking water of six countries (Germany, Australia, France, South Africa, the Netherlands and Spain), in order to assess the potential risks for ecology and human health. Atenolol was detected in all samples of the six countries under study. The highest atenolol concentrations were found in WWTP effluents, and ranged from 0.2 µg/L in Germany and in the Netherlands to 1.3 µg/L in Australia.
β-blockers were also found in several other Europea countries. In Sweden, atenolol, metoprolol, and propranolol were detected in the Hoje River at concentrations between 10 and 60 ng/L [172]. In Finland, Vieno et al. [63] found β-blockers in the effluents of three WWTPs at levels ranging from 910 to 1070 ng/L for metoprolol and from 40 to 440 ng/L for atenolol. The presence and fate of four β-blockers in wastewater and surface water in Switzerland was also reported [10]. The β-blocker concentrations in the three treatment plants of Dubendorf, Kloten-Opfikon and Niederglatt varied between 1980 and 2290 ng/L for atenolol, 290 and 400 ng/L for sotalol, and 240 and 260 ng/L for metoprolol in primary effluent.
The β-blocker concentrations were lower and ranged from 5 to 83 ng/L, in the Glatt River (Switzerland), and, in addition, four β-blockers, i.e. atenolol, metoprolol, propranol and sotalol, were found at low concentrations, ranging from 1 to 11 ng/L in drinking water in Luissane (Switzerland) by Morasch et al. [173]. In Poland, beta- blockers, such as sotalol, metoprolol and bisoprolol, were also reported at concentrations around 15 ng/L in tap water [174]. In Turkey, Aydin and Talinli. [175] found that the lake Buyukcekmece was contaminated by atenolol, and that WWTPs were characterized by the presence of atenolol and propranolol at concentrations ranging, respectively, from 2.7 to 48.5 ng/L and from 19.3 to 561 ng/L.
The presence of β-blockers was also reported in North America. Huggett et al. [35] performed the first study on the contamination by β-blockers of municipal wastewater effluent samples, collected from treatment facilities in Mississippi, Texas, and New York (USA), to determine the presence of metoprolol, nadolol, and propranolol. Propranolol was found in all wastewater samples (n = 34) at a concentration of 1.9 µg/L, as well as metoprolol and nadolol at concentrations of 1.2 µg/L and 0.36 µg/L, respectively. Batt et al. [176] reported the presence of atenolol, metoprolol and propranolol in wastewater from WWTPs and surface waters in the USA, at concentrations of 0.96 µg/L, 0.65 µg/L and 0.077 µg/L, respectively, in effluents. Similarly, the presence of metoprolol and propranolol was identified in more than fifty treatment plants of the USA [177]. An atenolol concentration of 3 µg/L was found by Cantwell et al. [178], who studied the temporal and spatial behavior of β-blockers in samples from the Narragansett Bay (USA). Metoprolol concentrations between 1.1 and 313 ng/L were determined, with highest levels obtained during the summer. Cantwell et al. [179] also found metoprolol in the Transect River, at concentrations ranging from 8 to 2020 ng/L. Oliveira et al. [180] realized a study on the fate of one hundred and eighty-five pharmaceuticals, including nine β-blockers, in effluents of hospitals and WWTPs. Metoprolol had the highest concentration of 3.54 μg/L in the USA.
The existence of β-blockers was also shown in Canada environmental waters. For example, Lee et al. [49] quantified β-blockers in municipal water treatment plants, and an atenolol highest concentration of 1650 ng/L was detected in all samples. Nadolol, acebutolol, sotalol, metoprolol, bisoprolol and propranolol were also found in all samples at lower concentrations. Similar results were obtained by MacLeod et al. [65], who quantified five β-blockers, among which atenolol was the most abundant, with levels of 971 ng/L and 664 ng/L, respectively, in influents and effluents. However, pindolol was not found in these samples. Recently, atenolol was detected in surface water and drinking water samples collected in the Ontario region [181]. An atenolol concentration of 6.9 ng/L was found in drinking water, about 5.6 times lower than that found in surface water.
β-blockers were also found in seawater, atenolol and metoprolol being the most important drugs. Nödler et al. [182] monitored pharmaceuticals in the coastal waters of different countries, with one hundred and fifty-three samples collected and analyzed in the Baltic sea coasts (Germany), the northern Adriatic Sea (Italy), the Aegean Sea, the Dardanelles (Greece and Turkey), the San Francisco Bay (USA), the Pacific Ocean (USA), the Mediterranean Sea (Israel) and the Balearic Sea (Spain). Atenolol, metoprolol and sotalol were detected in seawater from Germany, Greece, Turkey and Italy at concentrations ranging from 6 to 194 ng/L.
The presence of β-blockers was poorly documented in the African environmental waters, few studies being performed on the contamination of waters, mainly in South Africa. For example, Agunbiade and Moodley. [23] reported a high atenolol concentration of 39 µg/L in the South Africa influents. It was found that, after chlorination treatment, atenolol had a relatively high concentration. Moreover, Archer et al. [183] measured lower atenolol concentrations in the sewage treatment plan of the Gauteng province (South Africa). Values of 0.27 μg/L were found in the downstream of the river surface water, and of 0.156 μg/Lin the upstream. Pindolol was also detected in the influents and the effluents of the South African Daspoort wastewater treatment plant (Pretoria), at concentrations of 34.26 µg/L and 0.028 µg/L, respectively [184]. Metoprolol and pindolol were found at concentrations of 0.029 and 0.1 μg/L, respectively, in water samples of the Apies River, received from the Daspoort treatment plant. Atenolol was also detected in the surface waters of Yaoundé (Cameroon) with maximum concentrations of 0.016 µg/L in urban areas and of 0.005 µg/L in peri- urban areas [11]. Metoprolol and propranolol were found in Egypt, in effluents at maximum concentrations of 1.089 µg/L and 0.187 µg/L, respectively [185]. In the surface waters of Assiut, the maximum concentrations were 0.017 µg/L for metoprolol and 0.007 µg/L for propranolol. Ebele et al. [186] obtained similar results for metoprolol and propranolol at concentrations of 0.168 and 0.012 µg/L, respectively, in the Lagos surface waters (Nigeria). In addition, metoprolol was detected in drinking water at a concentration of 0.004 µg/L. The presence of atenolol and metoprolol was also reported at concentrations between 0.3 and 0.9 μg/L in the Meliane River (Tunisia), which received discharge from a wastewater treatment plant [187]. Atenolol was detected at maximum concentrations of 2.2 µg/L in influents and 1.2 µg/L in effluents [188]. Recently, Afsa et al. [189] measured atenolol concentrations up to 12.9 μg/L in hospital effluents.
In Asia, research performed in China, Japan and India showed the increased release of β-blockers in aquatic environment, due to the relocation of pharmaceutical industries in these countries, and to the lack of wastewater treatment infrastructure. For example, an Indian study of effluents from ninety commercial drug production facilities indicated the presence of the highest β-blocker concentrations worldwide [190], very high metoprolol concentrations ranging from 800 to 950 µg/L being found. Also, high atenolol concentrations, around 300 µg/L, were detected in India, due to the presence of important pharmaceutical industries. The presence of metoprolol was confirmed in the Ganges River with a maximum concentration of 24.4 ng/L [191]. Previous studies reported high metoprolol concentrations of 7 µg/L in lake waters and of 0.240 µg/L and 0.090 µg/L in river and well waters, respectively [192]. In Pakistan, atenolol, metoprolol and propranolol were detected in samples from drainage water, atenolol having the highest mean concentration (428 ng/L), followed by metoprolol (312 ng/L) [193].
In China, β-blockers such as atenolol, metoprolol and propranolol, were found in influents, with maximum concentrations of 995, 3665 and 3.6 ng/L respectively [194]. Moreover, these β-blockers were also detected in digested sludge at concentrations between 3.8 ng/g for propranolol and 17.9 ng/g for metoprolol. Xu et al.[6] reported the presence of five β-blockers in hospital wastewater, municipal wastewater, water bodies and agricultural land in Tianjin. β-blocker concentrations being higher in the hospital wastewater (10 µg/L) than in the municipal wastewater (5.2 µg/L). Duan et al. [195] reported the presence of metropolol concentrations between 8.15 and 1940 ng/L in urban and suburban areas of the Beiyun River Basin in Beijing. Recently, metoprolol concentrations of about 114.3 ng/L were also detected in the Chinese basin of Lake Tai [196]. In Japan, atenolol concentrations around 364 ng/L were found in wastewater. In Korea, atenolol and metoprolol were quantified in five wastewater treatment plants (WWTPs) in Ulsan, the largest countuy industrial city. Maximum concentrations of atenolol were greater than 10 µg/L in influent and 5.9 µg/L in effluent, while metoprolol lower concentrations were noted [197]. Also, lower concentrations of atenolol round 2.9 µg/L were detected in effluents in Taiwan [198].
Bioaccumulation in Aquatic Species, Plants and Food of Animal Origin
Aquatic species living in waters contaminated with pharmaceuticals were often exposed to these substances and could absorb them directly from water through respiration. For example, Wille et al. [87] reported the presence of β-blockers in marine organisms, namely blue mussels. Propranolol was detected at concentrations of 63 ng/g in blue mussels collected from Belgian coastal areas. Likewise, Huerta et al. [29] found the β-blockers carazolol and propranolol in fish from Mediterranean rivers at concentrations of 3.8 and 4.2 ng/g respectively. In another study, higher concentrations of propranolol (53 ng/g) were found in mussels, and an atenolol maximum concentration of 13 ng/g was detected in algae [199]. Atenolol was also present at concentrations of 0.3 ng/g in ribbed mussels collected from San Francisco Bay [200], and concentrations of 13 ng/g were also observed in mussels from the California coast [201].
The presence of atenolol was also reported at 62.7 ng/g in fish and 1.7 ng/g in shellfish collected from the Saudi Red Sea [202]. Research by Moreno-González et al. [28] revealed the presence of six β-blockers in different aquatic species at concentrations ranging from 0.5 to 1.5 ng/g in fish [73]. Reported the presence of atenolol, metoprolol and propranolol in oral species (common clam) at maximum concentrations of 2.7, 1.6 and 3 ng/g respectively. Recently, a review of Ruan et al. [205,206] revealed the chiral bioaccumulation of atenolol and metoprolol in twenty four marine species (five crustaceans, four mussels and fifteen fish). These β-blockers were present in all samples at concentrations ranging from 1.2 to 21.1 ng/g in crustaceans, from 0.02 to 1.98 ng/g in mussels, and from 0.02 to 4.67 ng/g in fish. A maximum bisoprolol concentration of 15 ng/g was detected in aquatic macroinvertebrates collected from the Fyris River in Uppsala (Sweden) [25].
The reuse of wastewater and the application of activated sludge as fertilizer in agricultural land could introduce several types of pollutants into the agroecosystems and, consequently, might contaminate plants. For instance, the presence of pharmaceutical compounds was reported in certain plant cultures (tomatoes, lettuces, carrots, etc.). Sabourin et al. [113]) noted the presence of atenolol at concentrations of 3.95 ng/g in tomatoes, and of 0.5 ng/g in potatoes. Recently, atenolol was also found at high concentrations of 850 ng/g in zucchini, and metoprolol was detected at a concentration of 105 ng/g in spinach [26].
Also, due to their illegal use in animal husbandry, the presence of β-blockers was investigated in animal meat, liver, muscle or milk. The most frequently detected β-blockers in these matrices were carazolol and metoprolol. For example, Delahaut et al. [203] followed the evolution of the carazolol concentration in the muscles, liver and kidneys of slaughtered pigs, 2, 6 and 24 h after administration of 0.25 mg/10 kg of carazolol. The highest concentrations of carazolol were observed after 2 h in all samples, ranging from 2 to 8 ppb. Similarly, Zhang et al. [96] detected metoprolol at a concentration of 3.5 µg/kg in pork muscle. However, recent studies on monitoring β-blockers in animal products did not detect any β-blockers, probably because of the strict regulations in place [72-78].
CONCLUSION
Pharmaceutical compounds present in the environment were considered as emerging contaminants, due to their persistence and bioaccumulation characteristics that might yield risks to the ecosystem and the human health. Several researches were performed to detect these micropollutants in environmental waters, and to propose effective treatment methods for their elimination. We provided here a comprehensive review on β-blockers, which were prescribed as pharmaceuticals for the treatment of different cardiovascular diseases. The present study focused on the presence of β-blockers in various environmental compartments, as well as on the development of analytical methods for their detection.
According to the literature, the presence of β-blockers was found in practically all environmental compartments, such as WWTP wastewaters, surface waters, groundwaters, tap waters, soils and other environmental matrices at small concentrations in the ng/mL to µg/mL range. In addition, their bioaccumulation in certain aquatic species, in plants (tomato, lettuce, etc.) and in food of animal origin (meat, liver, milk) was confirmed. Direct discharges of urban wastewater or WWTPs were found to be the main emission routes, but discharges from hospitals and pharmaceutical industries were also important. Most research results came from industrialized countries, but few studies were presented in Africa. The contamination of environmental matrices by β-blockers was found to be generally higher in African waters than on other continents, which indicated the lack of environmental data and of specialized laboratories in Africa (except South Africa) is therefore probably not due to lower pollution levels, but to the lack of equipped and funded to monitor pharmaceutical products in environmental matrices.
Major technological advances in the selectivity and specificity of analytical methods were demonstrated in the literature. However, because of the sample complexity, cleaning and preconcentration steps were necessary to eliminate possible interfering species before analysis. For the extraction of β-blockers from water samples, the Solid Phase Extraction (SPE) procedure, with the use of Oasis HLB or Oasis MCX cartridges, was found to be the most effective procedure, making possible to achieve satisfactory recoveries. Currently, Gas Chromatography (GC) and High- Performance Liquid Chromatography (HPLC) were the most applied analytical methods for the determination of pharmaceuticals, due to their high selectivity and sensitivity. However, since β-blockers are very polar, HPLC was the most appropriate method for their determination due to the incomplete derivatization in GC-MS. However, the latter method could also be effective with appropriate derivatization of β-blockers, despite their low volatility. Thanks to chromatographic approaches, such as HPLC coupled with different detection techniques like MS or MS/ MS, UV-DAD and FD, the identification and quantification of β-blockers could be performed, with low LOD and LOQ values in various matrices.
Spectrofluorimetry was also used for the analysis of β-blockers in biological samples and pharmaceutical formulations, since most β-blockers were naturally fluorescent. This method proved to be very selective and very sensitive, and it allowed the quantification of β-blockers formulations with LOD and LOQ values in the order of ng mL-1. Its performance was significantly improved with the use of cyclodextrins or micelles, due to the formation of inclusion complexes which enhanced the fluorescence signal. Additionally, for the simultaneous analysis of pharmaceutical compounds in binary or multi-component mixtures, synchronous fluorescence was very efficient for their separation and detection of β-blockers. Consequently, simple and synchronous spectrofluorimetry methods might be an alternative for the routine monitoring of pharmaceutical products at trace levels in the environment, filling the analytical gap existing between Africa and the other continents.
ACKNOWLEDGEMENTS
One of us (C. Gueye) thanks the Service of Cooperation and Cultural Action (SAC) of the French Embassy in Dakar (Senegal) for a PhD grant.
REFERENCES
- Frishman W.H. The beta-adrenoceptor blocking drugs. International Journal of Cardiology. 1982; 2: 165-178.
- Gabet-Giraud V, Miège C, Jacquet R, Coquery M. Impact of wastewater treatment plants on receiving surface waters and a tentative risk evaluation: the case of estrogens and beta blockers. Environ Sci Pollut Res. 2014; 21: 1708-1722.
- Ramil M, El Aref T, Fink G, Scheurer M, Ternes T.A. Fate of Beta Blockers in Aquatic-Sediment Systems: Sorption and Biotransformation. Environ Sci Technol. 2010; 44: 962-970.
- Uzelac M.M, Armakovi? S.J, Armakovi? S, ?etojevi?-Simin D.D, Agbaba J, Bani? N.D.. The role of environmental waters ionic composition and UV–LED radiation on photodegradation, mineralization and toxicity of commonly used β-blockers. Journal of Molecular Structure. 2022.
- Moore C.L, Henry D.S, McClenahan S.J, Ball K.K, Rusch N.J, Rhee S.W. Metoprolol Impairs β 1-Adrenergic Receptor-Mediated Vasodilation in Rat Cerebral Arteries: Implications for β -Blocker Therapy. J Pharmacol Exp Ther. 2021; 376: 127-135.
- Xu J, Sun H, Zhang Y, Alder A.C. Occurrence and enantiomer profiles of β-blockers in wastewater and a receiving water body and adjacent soil in Tianjin, China. Sci Total Environ. 2019; 650: 1122-1130.
- Salem A.A, Wasfi I.A, Al-Nassibi S.S. Trace determination of β-blockers and β2-agonists in distilled and waste-waters using liquid chromatography–tandem mass spectrometry and solid-phase extraction. Journal of Chromatography B. 2012; 908: 27-38.
- Al-Qaim F.F, Mussa Z.H, Yuzir A. Development and validation of a comprehensive solid-phase extraction method followed by LC-TOF/ MS for the analysis of eighteen pharmaceuticals in influent and effluent of sewage treatment plants. Anal Bioanal Chem. 2018; 410: 4829-4846.
- Shaheen J.F, Sizirici B, Yildiz, I. Fate, transport, and risk assessment of widely prescribed pharmaceuticals in terrestrial and aquatic systems: A review. Emerging Contaminants. 2022; 8: 216-228.
- Alder A.C, Schaffner C, Majewsky M, Klasmeier J, Fenner, K. Fate of β-blocker human pharmaceuticals in surface water: Comparison of measured and simulated concentrations in the Glatt Valley Watershed, Switzerland. Water Research. 2010; 44: 936-948.
- Branchet P, Ariza Castro N, Fenet H, Gomez E, Courant F, Sebag, D. et.al. Anthropic impacts on Sub-Saharan urban water resources through their pharmaceutical contamination (Yaoundé, Center Region, Cameroon). Sci Total Environ. 2019; 660: 886-898.
- Hernando M.D, Petrovic M, Fernández-Alba A.R, Barceló, D. Analysis by liquid chromatography–electrospray ionization tandem mass spectrometry and acute toxicity evaluation for β-blockers and lipid- regulating agents in wastewater samples. J Chromatogr A. 2004; 1046: 133-140.
- Jaukovi? Z.D, Gruji? S.D, Vasiljevi? T.M, Petrovi? S.D, Lauševi? M.D. Cardiovascular Drugs in Environmental Waters and Wastewaters: Method Optimization and Real Sample Analysis. J AOAC Int. 2014; 97: 1167-1174.
- Kanama K.M, Daso A.P, Mpenyana-Monyatsi L, Coetzee M.A.A. Assessment of Pharmaceuticals, Personal Care Products, and Hormones in Wastewater Treatment Plants Receiving Inflows from Health Facilities in North West Province, South Africa. Journal of Toxicology. 2018; 1-15.
- Kibbey T.C.G, Paruchuri R, Sabatini D.A, Chen L. 2007. Adsorption of Beta Blockers to Environmental Surfaces. Environ. Sci. Technol. 2007; 41: 5349-5356.
- Li Q, Jing S, Zhang J, Zhang L, Ran C, Du, C. et.al. Hollow fiber-protected liquid-phase microextraction followed by high performance liquid chromatography for simultaneously screening multiple trace level β-blockers in environmental water samples. Anal. Methods. 2015; 7: 6251-6259.
- Miège C, Favier M, Brosse C, Canler J-P, Coquery, M. 2006. Occurrence of betablockers in effluents of wastewater treatment plants from the Lyon area (France) and risk assessment for the downstream rivers. Talanta. 2006; 70: 739-744.
- Sanganyado E, Fu Q, Gan J. 2016. Enantiomeric selectivity in adsorption of chiral β-blockers on sludge. Environmental Pollution. 2016; 214: 787-794.
- Ternes T.A, Hirsch R, Mueller J, Haberer K. Methods for the determination of neutral drugs as well as betablockers and β 2-sympathomimetics in aqueous matrices using GC/MS and LC/MS/ MS. Fresenius’ Journal of Analytical Chemistry. 1998; 362: 329-340.
- Wang Z, Zhang X, Jiang S, Guo X. Magnetic solid-phase extraction based on magnetic multiwalled carbon nanotubes for the simultaneous enantiomeric analysis of five β-blockers in the environmental samples by chiral liquid chromatography coupled with tandem mass spectrometry. Talanta. 2018; 180: 98-107.
- Silva M, Morante-Zarcero S, Pérez-Quintanilla D, Sierra I. Simultaneous determination of pindolol, acebutolol and metoprolol in waters by differential-pulse voltammetry using an efficient sensor based on carbon paste electrode modified with amino-functionalized mesostructured silica. Sensors and Actuators B: Chemical. 2019; 283: 434-442.
- Yi M, Sheng Q, Sui Q, Lu H. β-blockers in the environment: Distribution, transformation, and ecotoxicity. Environ Pollut. 2020; 266: 115269.
- Agunbiade F.O, Moodley B. Pharmaceuticals as emerging organic contaminants in Umgeni River water system, KwaZulu-Natal, South Africa. Environ Monit Assess. 2014; 186: 7273-7291.
- Ogunbanwo O.M, Kay P, Boxall A.B, Wilkinson J, Sinclair C.J, Shabi, R.A. et.al. High Concentrations of Pharmaceuticals in a Nigerian River Catchment. Environ Toxicol Chem etc. 2020; 41: 551-558.
- Sedvall E, Fick J, Pettersson C, Hedeland M. Pharmaceuticals are identified in insects in River Fyris – A study with both tandem quadrupole and quadrupole-time-of-flight mass spectrometry. Environmental Advances. 2022: 8: 100194.
- Mansilla S, Portugal J, Bayona J.M, Matamoros V, Leiva A.M, Vidal G. et.al. Compounds of emerging concern as new plant stressors linked to water reuse and biosolid application in agriculture. Journal of Environmental Chemical Engineering. 2021; 9: 105198.
- Maskrey B.H, Dean K, Morrell N, Turner A.D. A Simple and Rapid Ultra–High-Performance Liquid Chromatography–Tandem Mass Spectrometry Method for the Quantitation of Pharmaceuticals and Related Compounds in Mussels and Oysters. Environ Toxicol Chem.2021; 40: 3263-3274.
- Moreno-González R, Rodríguez-Mozaz S, Huerta B, Barceló, D, León V.M. Do pharmaceuticals bioaccumulate in marine molluscs and fish from a coastal lagoon? Environmental Research. 2016; 146: 282-298.
- Huerta B, Jakimska A, Gros M, Rodríguez-Mozaz S, Barceló D. Analysis of multi-class pharmaceuticals in fish tissues by ultra-high- performance liquid chromatography tandem mass spectrometry. Journal of Chromatography A. 2013; 1288: 63-72.
- Changotra R, Rajput H, Dhir A. Treatment of real pharmaceutical wastewater using combined approach of Fenton applications and aerobic biological treatment. Journal of Photochemistry and Photobiology A: Chemistry. 2019; 376: 175-184.
- Xiao X, Zhang M-M, Wang Z-Q. Determination of β-Blockers in Bovine and Porcine Tissues and Bovine Milk by High-Performance Liquid Chromatography – Tandem Mass Spectrometry. Analytical Letters. 2019; 52: 439-451.
- Cleuvers M. Initial risk assessment for three β-blockers found in the aquatic environment. Chemosphere. 2005; 59: 199-205.
- Di Lorenzo T, Castaño-Sánchez A, Di Marzio W.D, García-Doncel P, Nozal Martínez, L, Galassi D.M.P. et.al. The role of freshwater copepods in the environmental risk assessment of caffeine and propranolol mixtures in the surface water bodies of Spain. Chemosphere. 2019; 220: 227-236.
- Godoy A.A, Kummrow F, Pamplin P.A.Z. Occurrence, ecotoxicological effects and risk assessment of antihypertensive pharmaceutical residues in the aquatic environment - A review. Chemosphere. 2015; 138: 281-291.
- Huggett D.B, Brooks B.W, Peterson B, Foran C.M, Schlenk D. Toxicity of Select Beta Adrenergic Receptor-Blocking Pharmaceuticals (B-Blockers) on Aquatic Organisms. Archives of Environmental Contamination and Toxicology. 2002; 43: 229-235.
- Maszkowska J, Stolte S, Kumirska J, ?ukaszewicz P, Mioduszewska K, Puckowski, A. et.al. 2014. Beta-blockers in the environment: Part II. Ecotoxicity study. Science of The Total Environment. 2014; 493: 1122–1126.
- Massarsky A, Trudeau V.L, Moon T.W. β-blockers as endocrine disruptors: the potential effects of human β-blockers on aquatic organisms. J. Exp. Zool A Ecol Genet Physol. 2011; 315: 251–265.
- Médicaments et environnement 59. Académie nationale de pharmacie. 2019.
- Wada 2021, n.d.
- Cheng J-Q, Liu T, Nie X-M, Chen F-M, Wang C-S, Zhang F. Analysis of 27 β-Blockers and Metabolites in Milk Powder by High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap High- Resolution Mass Spectrometry. Molecules. 2019; 24: 820.
- Mitrowska K, Posyniak A, Zmudzki J. Rapid method for the determination of tranquilizers and a beta-blocker in porcine and bovine kidney by liquid chromatography with tandem mass spectrometry. Anal Chimi Acta. 2009; 637: 185-192.
- Basheer C, Wang H, Jayaraman A, Valiyaveettil S, Lee H.K. Polymer- coated hollow fiber microextraction combined with on-column stacking in capillary electrophoresis. Journal of Chromatography A. 2006; 1128: 267-272.
- Silva M, Morante-Zarcero S, Pérez-Quintanilla D, Marina M.L, SierraI. Preconcentration of β-blockers using functionalized ordered mesoporous silica as sorbent for SPE and their determination in waters by chiral CE. ELECTROPHORESIS. 2017; 38: 1905-1912.
- Basan H, Yar?mkaya S. A novel solid-phase extraction- spectrofluorimetric method for the direct determination of atenolol in human urine: A novel spectrofluoimetriv method for the determination of atenolol. Luminescence. 2014; 29: 225-229.
- Gorbani Y, Y?lmaz H, Basan H. Spectrofluorimetric determination of atenolol from human urine using high-affinity molecularly imprinted solid-phase extraction sorbent. Luminescence.2017; 32: 1391-1397.
- Gueye C, Aaron J-J, Gaye-Seye M.D, Cisse L, Oturan N, Oturan M.A. A spectrofluorimetric method for the determination of pindolol in natural waters using various organic and cyclodextrin media. Environ Sci Pollut Res Int. 2021; 28: 55029-55040.
- Madrakian T, Afkhami A, Mohammadnejad M. Simultaneous spectrofluorimetric determination of levodopa and propranolol in urine using feed-forward neural networks assisted by principal component analysis. Talanta. 2009; 78: 1051-1055.
- Y?ld?r?m S, Erkmen C, Uslu B. Novel Trends in Analytical Methods for β-Blockers: An Overview of Applications in the Last Decade. Crit Rev Anal Chem. 2020; 52: 131-169.
- Lee H-B, Sarafin K, Peart T.E. Determination of β-blockers and β2-agonists in sewage by solid-phase extraction and liquid chromatography–tandem mass spectrometry. Journal of Chromatography A. 2007; 1148: 158-167.
- Mackay D, Arnot JA, Gobas FA, Powell DE. Mathematical relationships between metrics of chemical bioaccumulation in fish. Environ Toxicol Chem. 2013; 32: 1459-1466.
- M. Dolores Hernando, M. José Gómez, Ana Agüera, Amadeo R. Fernández-Alba. LC-MS analysis of basic pharmaceuticals (beta- blockers and anti-ulcer agents) in wastewater and surface water, TrAC Trends in Analytical Chemistry. 2007; 26: 581-594.
- Zhang K, Zhao Y, Fent K. Cardiovascular drugs and lipid regulating agents in surface waters at global scale: Occurrence, ecotoxicity and risk assessment. Sci Total Environ. 2020; 729: 138770.
- Braza AJ, Modamio P, Lastra CF, Mariño EL. Development, validation and analytical error function of two chromatographic methods with fluorimetric detection for the determination of bisoprolol and metoprolol in human plasma. Biomed Chromatogr. 2002; 8: 517-522.
- Kadam RS, Kompella UB. Cassette analysis of eight beta-blockers in bovine eye sclera, choroid-RPE, retina, and vitreous by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009; 877: 253-260.
- Yilmaz B, Arslan S. Determination of atenolol in human urine by using HPLC. Sep Sci plus 2018; 1: 4-10.
- Tomková J, Ondra P, Kocianová E, Václavík J. Fast and sensitive analysis of beta blockers by ultra-high-performance liquid chromatography coupled with ultra-high-resolution TOF mass spectrometry. Biomed Chromatogr. 2017; 31.
- Andrade-Eiroa A, Canle M, Leroy-Cancellieri V, Cerdà V. Solid-phase extraction of organic compounds: A critical review (Part I). TrAC Trends in Analytical Chemistry. 2016; 80: 641-654.
- Caban, Magda, Piotr Stepnowski, Marek Kwiatkowski, Joanna Maszkowska, Marta Wagil and Jolanta Kumirska. Comparison of the Usefulness of SPE Cartridges for the Determination of β-Blockers and β-Agonists (Basic Drugs) in Environmental Aqueous Samples. Journal of Chemistry. 2015; 2015: 1-9.
- Gros M, Petrovi? M, Barceló D. Development of a multi-residue analytical methodology based on liquid chromatography-tandem mass spectrometry (LC-MS/MS) for screening and trace level determination of pharmaceuticals in surface and wastewaters. Talanta. 2006; 704: 678-690.
- Josefsson M, Sabanovic A. Sample preparation on polymeric solid phase extraction sorbents for liquid chromatographic-tandem mass spectrometric analysis of human whole blood--a study on a number of beta-agonists and beta-antagonists. J Chromatogr A. 2006; 1120: 1-12.
- Gros M, Pizzolato TM, Petrovi? M, de Alda MJ, Barceló D. Trace level determination of beta-blockers in waste waters by highly selective molecularly imprinted polymers extraction followed by liquid chromatography-quadrupole-linear ion trap mass spectrometry. J Chromatogr A. 2008; 1189: 374-384.
- Caban M, Lis E, Kumirska J, Stepnowski P. Determination of pharmaceutical residues in drinking water in Poland using a new SPE-GC-MS(SIM) method based on Speedisk extraction disks and DIMETRIS derivatization. Sci Total Environ. 2015; 538: 402-411.
- Vieno NM, Tuhkanen T, Kronberg L. Analysis of neutral and basic pharmaceuticals in sewage treatment plants and in recipient rivers using solid phase extraction and liquid chromatography-tandem mass spectrometry detection. J Chromatogr A. 2006; 1134: 101-111.
- Nikolai LN, McClure EL, Macleod SL, Wong CS. Stereoisomer quantification of the beta-blocker drugs atenolol, metoprolol, and propranolol in wastewaters by chiral high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2006; 1131: 103-109.
- MacLeod SL, Sudhir P, Wong CS. Stereoisomer analysis of wastewater- derived beta-blockers, selective serotonin re-uptake inhibitors, and salbutamol by high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2007; 1170: 23-33.
- Wu Y, Fan S, Zhao Y, Miao H. Determination of 23 β2-agonists and 5 β-blockers in animal muscle by high performance liquid chromatography-linear ion trap mass spectrometry. Science China Chemistry. 2010; 53: 832-840.
- Jakubus, Aleksandra, Maciej Gromelski, Karolina Jagiello, Tomasz Puzyn, Piotr Stepnowski, and Monika Paszkiewicz. Dispersive Solid- Phase Extraction Using Multi-Walled Carbon Nanotubes Combined With Liquid chromatography–mass Spectrometry for the Analysis of β-Blockers: Experimental and Theoretical Studies. Microchemical Journal. 2019; 146: 258-269.
- de Oliveira LG, Barreto F, Hoff R, Rübensam G, Scherer Kurz MH, Galle G, Gonçalves FF. Validation of a method for sedatives and β-blockers determination in swine, bovine and equine kidney using liquid chromatography coupled with tandem mass spectrometry. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2017; 34: 32-39.
- Parrilla Vázquez MM, Parrilla Vázquez P, Martínez Galera M, Gil García MD, Uclés A. Ultrasound-assisted ionic liquid dispersive liquid-liquid microextraction coupled with liquid chromatography-quadrupole- linear ion trap-mass spectrometry for simultaneous analysis of pharmaceuticals in wastewaters. J Chromatogr A. 2013; 1291: 19-26.
- López-Serna R, Petrovi? M, Barceló D. Direct analysis of pharmaceuticals, their metabolites and transformation products in environmental waters using on-line TurboFlow™ chromatography- liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2012; 1252: 115-129.
- López-Serna R, Pérez S, Ginebreda A, Petrovi? M, Barceló D. Fully automated determination of 74 pharmaceuticals in environmental and waste waters by online solid phase extraction-liquid chromatography-electrospray-tandem mass spectrometry. Talanta. 2010; 83: 410-424.
- Shang J, He X, Xi C, Tang B, Wang G, Chen D, Peng T, Mu Z. Determination of the potential illegal addition of β-blockers to function foods by QuEChERS sample preparation and UPLC-MS/MS analysis. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2015; 32: 1040-1048.
- Rodrigues J, Albino S, Silva S, Cravo A, Cardoso VV, Benoliel MJ, et al. Development of a Multiresidue Method for the Determination of 24 Pharmaceuticals in Clams by QuEChERS and Liquid Chromatography- Triple Quadrupole Tandem Mass Spectrometry. Food Anal. Methods. 2019; 12: 838-851.
- Ramirez AJ, Mottaleb MA, Brooks BW, Chambliss CK. Analysis of pharmaceuticals in fish using liquid chromatography-tandem mass spectrometry. Anal Chem. 2007; 79: 3155-3163.
- Šrámková, Ivana H. et al. “On-line coupling of Micro-Extraction by Packed Sorbent with Sequential Injection Chromatography system for direct extraction and determination of betaxolol in human urine.” Talanta. 2015; 143: 132-137.
- Pereira, J.A.M., Gonçalves, J., Porto-Figueira, P., Figueira, J.A., Alves, V., Perestrelo, R.,et al. Current trends on microextraction by packed sorbent – fundamentals, application fields, innovative improvements and future applications. Analyst. 2019; 144: 5048-5074.
- Mazaraki K, Kabir A, Furton KG, Fytianos K, Samanidou VF, Zacharis CK. Fast fabric phase sorptive extraction of selected β-blockers from human serum and urine followed by UHPLC-ESI-MS/MS analysis. J Pharm Biomed Anal. 2021; 199: 114053.
- Wu, C., Ning, X., Chen, X., Ma, J., Zhao, Q., Zhao, L.,et al. Multi-functional porous organic polymers for highly-efficient solid-phase extraction of β-agonists and β-blockers in milk. RSC Adv. 2021; 46: 28925-28933.
- Iha MH, Martinez AS, Bonato PS. Enantioselective analysis of atenolol in biologic fluids: comparison of liquid-liquid and solid-phase extraction methods. J Chromatogr B Analyt Technol Biomed Life Sci. 2002; 767: 1-9.
- Kadam RS, Kompella UB. Cassette analysis of eight beta-blockers in bovine eye sclera, choroid-RPE, retina, and vitreous by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009; 877: 253-260.
- Aranas AT, Guidote AM Jr, Haddad PR, Quirino JP. Sweeping-micellar electrokinetic chromatography for the simultaneous analysis of tricyclic antidepressant and β-blocker drugs in wastewater. Talanta. 2011; 85: 86-90.
- Bilal Yilmaz, Sakir Arslan, Determination of Carvedilol in Human Plasma by Gas Chromatography-Mass Spectrometry Method. Journal of Chromatographic Science. 2011; 49: 35-39.
- Mazzarino M, Fiacco I, de la Torre X, Botrè F. Screening and confirmation analysis of stimulants, narcotics and beta-adrenergic agents in human urine by hydrophilic interaction liquid chromatography coupled to mass spectrometry. J Chromatogr A. 2011; 1218: 8156-8167.
- Giebu?towicz J, Kojro G, Bu?-Kwa?nik K, Rudzki PJ, Marsza?ek R, Le? A, et al. Cloud-point extraction is compatible with liquid chromatography coupled to electrospray ionization mass spectrometry for the determination of bisoprolol in human plasma. J Chromatogr A. 2015; 1423: 39-46.
- Wan Q, Liu Y, Yang C, Liu L. On-line double focusing of atenolol and metoprolol in human urine using capillary electrophoresis with the aid of β-cyclodextrin. Anal Chim Acta. 2017; 978: 61-67.
- Caban M, Migowska N, Stepnowski P, Kwiatkowski M, Kumirska J. Matrix effects and recovery calculations in analyses of pharmaceuticals based on the determination of β-blockers and β-agonists in environmental samples. J Chromatogr A. 2012; 1258: 117-1127.
- Wille, K., Kiebooms, J.A.L., Claessens, M., Rappé, K., Vanden Bussche, J., Noppe, H., et al. Development of analytical strategies using U-HPLC- MS/MS and LC-ToF-MS for the quantification of micropollutants in marine organisms. Anal Bioanal Chem. 2011; 400: 1459-1472.
- Saleem K, Ali I, Kulsum U, Aboul-Enein HY. Recent developments in HPLC analysis of β-blockers in biological samples. J Chromatogr Sci. 2013; 51: 807-818.
- Rivera-Jaimes JA, Postigo C, Melgoza-Alemán RM, Aceña J, Barceló D, López de Alda M. Study of pharmaceuticals in surface and wastewater from Cuernavaca, Morelos, Mexico: Occurrence and environmental risk assessment. Sci Total Environ. 2018; 613-614: 1263-1274.
- Stankiewicz A, Giebu?towicz J, Stankiewicz U, Wroczy?ski P, Na??cz-Jawecki G. Determination of selected cardiovascular active compounds in environmental aquatic samples--Methods and results, a review of global publications from the last 10 years. Chemosphere. 2015; 138: 642-656.
- Scheurer M, Ramil M, Metcalfe CD, Groh S, Ternes TA. The challenge of analyzing beta-blocker drugs in sludge and wastewater. Anal Bioanal Chem. 2010; 396: 845-856.
- De Nicolò A, Avataneo V, Rabbia F, Sciandra M, Tosello F, Cusato J, et al. UHPLC–MS/MS method with sample dilution to test therapeutic adherence through quantification of ten antihypertensive drugs in urine samples. Journal of Pharmaceutical and Biomedical Analysis. 2017; 142: 279-285.
- Gory?ski K, Kiedrowicz A, Bojko B. Development of SPME-LC-MS method for screening of eight beta-blockers and bronchodilators in plasma and urine samples. J Pharm Biomed Anal. 2016; 127: 147-155.
- Kristoffersen L, Øiestad EL, Opdal MS, Krogh M, Lundanes E, Christophersen AS. Simultaneous determination of 6 beta-blockers, 3 calcium-channel antagonists, 4 angiotensin-II antagonists and 1 antiarrhythmic drug in post-mortem whole blood by automated solid phase extraction and liquid chromatography mass spectrometry. Method development and robustness testing by experimental design. J Chromatogr B Analyt Technol Biomed Life Sci. 2007; 850: 147-160.
- Sai F, Hong M, Yunfeng Z, Huijing C, Yongning W. Simultaneous detection of residues of 25 β?-agonists and 23 β-blockers in animal foods by high-performance liquid chromatography coupled with linear ion trap mass spectrometry. J Agric Food Chem. 2012; 60: 1898-1905.
- Zhang J, Shao B, Yin J, Wu Y, Duan H. Simultaneous detection of residues of beta-adrenergic receptor blockers and sedatives in animal tissues by high-performance liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009; 877: 1915-1922.
- Gros M, Petrovi? M, Barceló D. Tracing pharmaceutical residues of different therapeutic classes in environmental waters by using liquid chromatography/quadrupole-linear ion trap mass spectrometry and automated library searching. Anal Chem. 2009; 81: 898-912.
- ?eli?, M., Farré, M., Lopez de Alda, M., Perez, S., Barceló, D., Petrovi?,M. Environmental analysis: Emerging pollutants, in: Liquid Chromatography. Elsevier. 2017; 451-477.
- Stankiewicz A, Giebu?towicz J, Stefa?ski M, Sikorska K, Wroczy?ski P, Na??cz-Jawecki G. The development of the LC-MS/MS method based on S-9 biotransformation for detection of metabolites of selected β-adrenolytics in surface water. Environ Toxicol Pharmacol. 2015; 39: 906-916.
- Abdel Hameed EA, Abdel Salam RA, Hadad GM. Chemometric- assisted spectrophotometric methods and high performance liquid chromatography for simultaneous determination of seven β-blockers in their pharmaceutical products: a comparative study. Spectrochim Acta A Mol Biomol Spectrosc. 2015; 141: 278-286
- Castiglioni S, Bagnati R, Calamari D, Fanelli R, Zuccato E. A multiresidue analytical method using solid-phase extraction and high-pressure liquid chromatography tandem mass spectrometry to measure pharmaceuticals of different therapeutic classes in urban wastewaters. J Chromatogr A. 2005; 1092: 206-215.
- Huggett DB, Khan IA, Foran CM, Schlenk D. Determination of beta- adrenergic receptor blocking pharmaceuticals in United States wastewater effluent. Environ Pollut. 2003; 121: 199-205.
- Mohammed, G.I., Metwally Nassef, H., Bashammakh, Abdulaziz.S., Saigl, Zainab.M., El-Shahawi, M.S. Analysis of β-blocker timolol maleate drug residues in wastewater and biological fluids using differential pulse – anodic stripping voltammetry. International Journal of Environmental Analytical Chemistry. 2020; 102: 1-17.
- Morante-Zarcero S, Sierra I. Comparative HPLC methods for β-blockers separation using different types of chiral stationary phases in normal phase and polar organic phase elution modes. Analysis of propranolol enantiomers in natural waters. J Pharm Biomed Anal. 2012; 62: 33-41.
- Valcárcel Y, Alonso SG, Rodríguez-Gil JL, Maroto RR, Gil A, Catalá M. Analysis of the presence of cardiovascular and analgesic/anti- inflammatory/antipyretic pharmaceuticals in river- and drinking- water of the Madrid Region in Spain. Chemosphere. 2011; 82: 1062- 1071.
- Parrilla Vázquez P, Martínez Galera M, Serrano Guirado A, Parrilla Vázquez MM. Determination of five beta-blockers in wastewaters by coupled-column liquid chromatography and fluorescence detection. Anal Chim Acta. 2010; 666: 38-44.
- Thomas A Ternes, Analytical methods for the determination of pharmaceuticals in aqueous environmental samples, TrAC Trends in Analytical Chemistry. 2001; 20: 419-434.
- Sacher F, Lange FT, Brauch HJ, Blankenhorn I. Pharmaceuticals in groundwaters analytical methods and results of a monitoring program in Baden-Württemberg, Germany. J Chromatogr A. 2001; 938: 199-210.
- Gros M, Rodríguez-Mozaz S, Barceló D. Fast and comprehensive multi-residue analysis of a broad range of human and veterinary pharmaceuticals and some of their metabolites in surface and treated waters by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap tandem mass spectrometry. J Chromatogr A. 2012; 1248: 104-121.
- Petrovi? M, Škrbi? B, Živan?ev J, Ferrando-Climent L, Barcelo D. Determination of 81 pharmaceutical drugs by high performance liquid chromatography coupled to mass spectrometry with hybrid triple quadrupole-linear ion trap in different types of water in Serbia. Sci Total Environ. 2014; 468-469: 415-428.
- Gómez MJ, Petrovi? M, Fernández-Alba AR, Barceló D. Determination of pharmaceuticals of various therapeutic classes by solid-phase extraction and liquid chromatography-tandem mass spectrometry analysis in hospital effluent wastewaters. J Chromatogr A. 2006; 1114: 224-233.
- Maurer M, Escher BI, Richle P, Schaffner C, Alder AC. Elimination of beta-blockers in sewage treatment plants. Water Res. 2007; 41: 1614-1622.
- Sabourin L, Duenk P, Bonte-Gelok S, Payne M, Lapen DR, Topp E. Uptake of pharmaceuticals, hormones and parabens into vegetables grown in soil fertilized with municipal biosolids. Sci Total Environ. 2012; 431: 233-226.
- Varga R, Somogyvári I, Eke Z, Torkos K. Seasonal Monitoring of Cardiovascular and Antiulcer Agents’ Concentrations in Stream Waters Encompassing a Capital City. J Pharm (Cairo). 2013; 2013: 753928.
- De Andrés F, Castañeda G, Ríos A. Use of toxicity assays for enantiomeric discrimination of pharmaceutical substances. Chirality. 2009; 21: 751-759.
- Sanganyado E, Lu Z, Fu Q, Schlenk D, Gan J. Chiral pharmaceuticals: A review on their environmental occurrence and fate processes. Water Res. 2017; 124: 527-542.
- Ribeiro, C., Ribeiro, A., Maia, A., Tiritan, M. Occurrence of Chiral Bioactive Compounds in the Aquatic Environment: A Review. 2017; 9: 215.
- Guo J, Wang J, Lin H, Feng Y, Shen H, Huang R, et al. Combination of capillary electrophoresis and molecular modeling to study the enantiomer affinity pattern between β-blockers and anionic cyclodextrin derivatives in a methanolic and water background electrolyte. J Sep Sci. 2019; 42: 1077-1087.
- P. Morin, Séparation de molécules pharmaceutiques chirales par les techniques chromatographiques et électrophorétiques. Annales Pharmaceutiques Françaises. 2009; 67: 241-250.
- Yang Y, Wang Y, Bao Z, Yang Q, Zhang Z, Ren Q. Progress in the Enantioseparation of β-Blockers by Chromatographic Methods. Molecules. 2021; 26: 468.
- Morante-Zarcero S, Sierra I. Simultaneous enantiomeric determination of propranolol, metoprolol, pindolol, and atenolol in natural waters by HPLC on new polysaccharide-based stationary phase using a highly selective molecularly imprinted polymer extraction. Chirality. 2012; 24: 860-866.
- Barclay VK, Tyrefors NL, Johansson IM, Pettersson CE. Chiral analysis of metoprolol and two of its metabolites, α-hydroxymetoprolol and deaminated metoprolol, in wastewater using liquid chromatography- tandem mass spectrometry. J Chromatogr A. 2012; 1269: 208-217.
- Svan A, Hedeland M, Arvidsson T, Jasper JT, Sedlak DL, Pettersson CE. Rapid chiral separation of atenolol, metoprolol, propranolol and the zwitterionic metoprolol acid using supercritical fluid chromatography-tandem mass spectrometry - Application to wetland microcosms. J Chromatogr A. 2015; 1409: 251-258.
- Arenas M, Santos JL, Martín J, Aparicio I, Alonso E. Enantioselective LC-MS/MS determination of antidepressants, β-blockers and metabolites in agricultural soil, compost and digested sewage sludge. Analytica Chimica Acta. 2023; 1261: 341224.
- Amendola L, Molaioni F, Botrè F. Detection of beta-blockers in human urine by GC-MS-MS-EI: perspectives for the antidoping control. J Pharm Biomed Anal. 2000; 23: 211-221.
- Pujos E, Cren-Olivé C, Paisse O, Flament-Waton MM, Grenier- Loustalot MF. Comparison of the analysis of beta-blockers by different techniques. J Chromatogr B Analyt Technol Biomed Life Sci. 2009; 877: 4007-4014.
- Santos MG, Tavares, IMC, Boralli, VB, Figueiredo EC. Direct doping analysis of beta-blocker drugs from urinary samples by on-line molecularly imprinted solid-phase extraction coupled to liquid chromatography/mass spectrometry. Analyst. 2015; 8: 2696-2703.
- Xu T, Bao S, Geng P, Luo J, Yu L, Pan P, et al. Determination of metoprolol and its two metabolites in human plasma and urine by high performance liquid chromatography with fluorescence detection and its application in pharmacokinetics. J Chromatogr B Analyt Technol Biomed Life Sci. 2013; 937: 60-66.
- Zhang Z, Yan H, Cui F, Yun H, Chang X, Li J, et al. Analysis of multiple β-agonist and β-blocker residues in porcine muscle using improved QuEChERS method and UHPLC-LTQ orbitrap mass spectrometry. Food Anal. Methods. 2016; 9: 915-924.
- Paik M-J, Nguyen DT, Kim K-R. GC and MS Properties of β-Blockers as tert-butyldimethylsilyl derivatives and as ethoxycarbonyl/ trimethylsilyl derivatives. Chroma 2006; 64: 673-679.
- Caban M, Stepnowski P, Kwiatkowski M, Migowska N, Kumirska J. Determination of β-blockers and β-agonists using gas chromatography and gas chromatography-mass spectrometry--a comparative study of the derivatization step. J Chromatogr A. 2011; 1218: 8110-8122.
- Caban M, Mioduszewska K, Stepnowski P, Kwiatkowski M, Kumirska J. Dimethyl(3,3,3-trifluoropropyl)silyldiethylamine--a new silylating agent for the derivatization of β-blockers and β-agonists in environmental samples. Anal Chim Acta. 2013; 782: 75-88.
- Fono LJ, Sedlak DL. Use of the chiral pharmaceutical propranolol to identify sewage discharges into surface waters. Environ Sci Technol. 2005; 39: 9244-9252.
- Jouyban A, Sorouraddin MH, Farajzadeh MA, Somi MH, Fazeli- Bakhtiyari R. Vortex-assisted liquid-liquid extraction combined with field-amplified sample injection and sweeping micellar electrokinetic chromatography for improved determination of β-blockers in human urine. Talanta. 2016; 149: 298-309.
- Al-Ghannam SM. A simple spectrophotometric method for the determination of beta-blockers in dosage forms. J Pharm Biomed Anal. 2006; 40: 151-156.
- Gölcü A. New, simple, and validated UV-spectrophotometric method for the estimation of some beta blockers in bulk and formulations. J Anal Chem. 2008; 63: 538-543.
- Vaikosen EN, Bioghele J, Worlu RC, Ebeshi, BU. Spectroscopic Determination of Two Beta-Blockers – Atenolol and Propanolol by Oxidative Derivatization Using Potassium Permanganate in Alkaline Medium. Reviews in Analytical Chemistry. 2020; 39: 56-64.
- Zakaria SA, Zakaria RA, Othman NS, Spectrophotometric determination of atenolol via oxidation and bleaching color reaction for methyl red dye. J. Phys.: Conf. Ser. 2021; 2063: 012008.
- Xu LX, Hui N, Ma LY, Wang HY. Study on fluorescence property of carvedilol and determination of carvedilol by fluorimetry. Spectrochim Acta A Mol Biomol Spectrosc. 2005; 61: 855-859.
- Zeeb M, Mirza B. Ionic liquid phase microextraction combined with fluorescence spectrometry for preconcentration and quantitation of carvedilol in pharmaceutical preparations and biological media. Daru. 2015; 23: 30.
- Silva RA, Talío MC, Luconi MO, Fernández LP. Evaluation of carbon nanotubes as chiral selectors for continuous-flow enantiomeric separation of carvedilol with fluorescent detection. J Pharm Biomed Anal. 2012; 70: 631-635.
- Ibrahim, FA, El-Brashy, AM, El-Awady, MI, Abdallah, Nora A. Development of a validated spectrofluorimetric method for assay of sotalol hydrochloride in tablets and human plasma: application for stability-indicating studies. Open Chemistry. 2019; 17: 64-74.
- Abdine H, Sultan MA, Hefnawy MM, Belal F. Spectrofluorometric determination of some beta-blockers in tablets and human plasma using 9,10-dimethoxyanthracene-2-sodium sulfonate. Pharmazie. 2005; 60: 265-268.
- Bakir E, Gouda M, Alnajjar A, Boraie WE. Spectrofluorimetric method for atenolol determination based on gold nanoparticles. Acta Pharm. 2018; 68: 243-250.
- Zhang H-M, Yang J-Y, Du L-M, Li C-F, Wu H. Determination of sotalol by fluorescence quenching method. Analytical Methods. 2011; 3: 1156-1162.
- Zhang F, Chen M, Zhang H, Xiong H, Wen W, Zhang X, Wang S. Fluorescence suppression of MPA stabilized CdTe QDs for direct determination of propranolol. Anal. Methods. 2017; 9: 929-936.
- Pérez Ruiz T, Martínez-Lozano C, Tomás V, Carpena J. Simultaneous determination of propranolol and pindolol by synchronous spectrofluorimetry. Talanta. 1998; 45: 969-976.
- Xiao Y, Wang HY, Han J. Simultaneous determination of carvedilol and ampicillin sodium by synchronous fluorimetry. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy. 2005; 61: 567-573.
- Walash MI, Belal FF, El-Enany NM, El-Maghrabey MH. Synchronous fluorescence spectrofluorimetric method for the simultaneous determination of metoprolol and felodipine in combined pharmaceutical preparation. Chemistry Central Journal. 2011; 5: 70.
- Mabrouk MM, Hammad SF, El-Malla SF, Elshenawy EA. Simultaneous determination of amlodipine and metoprolol in their combined dosage form using a synchronous fluorescence spectrofluorimetric method. Luminescence. 2018; 33: 364-369.
- Safwat N, Abdel-Ghany MF, Ayad MF. Sensitive Derivative Synchronous and Micellar Enhanced Ecofriendly Spectrofluorimetric Methods for the Determination of Atenolol, Diclofenac, and Triclosan in Drinking Tap Water. J AOAC Int. 2021; 104: 103-112.
- Abo Elkheir SM, Zeid AM, Nasr JJM, Walash MI. First derivative synchronous spectrofluorimetric analysis of bisoprolol fumarate and ivabradine in pharmaceutical and biological matrices. Investigation of the method greenness. Luminescence. 2022; 37: 1657-1665.
- Rezka P, Balcerzak W. Beta-adrenergic drugs (β-blockers) in the environment ? new methods of removal. 2015.
- Wick A, Fink G, Joss A, Siegrist H, Ternes TA. Fate of beta blockers and psycho-active drugs in conventional wastewater treatment. Water Res. 2009; 43: 1060-1074.
- Nödler K, Hillebrand O, Idzik K, Strathmann M, Schiperski F, Zirlewagen J, et al. Occurrence and fate of the angiotensin II receptor antagonist transformation product valsartan acid in the water cycle--a comparative study with selected β-blockers and the persistent anthropogenic wastewater indicators carbamazepine and acesulfame. Water Res. 2013; 47: 6650-6659.
- Gurke R, Rossmann J, Schubert S, Sandmann T, Rößler M, Oertel R, et al. Development of a SPE-HPLC-MS/MS method for the determination of most prescribed pharmaceuticals and related metabolites in urban sewage samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2015; 990: 23-30.
- Gabet-Giraud V, Miège C, Choubert JM, Ruel SM, Coquery M. Occurrence and removal of estrogens and beta blockers by various processes in wastewater treatment plants. Sci Total Environ. 2010; 408: 4257-4269.
- Jacquet R, Miège C, Bados P, Schiavone S, Coquery M. Evaluating the polar organic chemical integrative sampler for the monitoring of beta-blockers and hormones in wastewater treatment plant effluents and receiving surface waters. Environ Toxicol Chem. 2012; 31: 279- 288.
- Vulliet E, Cren-Olive C, Grenier-Loustalot MF. Occurrence of pharmaceuticals and hormones in drinking water treated from surface waters. Environ Chem Lett 12. 2011.
- Verlicchi P, Al Aukidy M, Galletti A, Petrovic M, Barceló D. Hospital effluent: Investigation of the concentrations and distribution of pharmaceuticals and environmental risk assessment. Sci Total Environ. 2012; 430: 109-118.
- Al Aukidy M, Verlicchi P, Jelic A, Petrovic M, Barcelò D. Monitoring release of pharmaceutical compounds: occurrence and environmental risk assessment of two WWTP effluents and their receiving bodies in the Po Valley, Italy. Sci Total Environ. 2012; 438: 15-25.
- Papagiannaki D, Morgillo S, Bocina G, Calza P, Binetti R. Occurrence and human health risk assessment of pharmaceuticals and hormones in drinking water sources in the metropolitan area of turin in Italy. Toxics. 2021; 9: 88.
- Muñoz I, López-Doval JC, Ricart M, Villagrasa M, Brix R, Geiszinger A, et al. Bridging levels of pharmaceuticals in river water with biological community structure in the Llobregat River basin (northeast Spain). Environ Toxicol Chem. 2009; 28: 2706-2714.
- Camacho-Muñoz D, Martín J, Santos JL, Aparicio I, Alonso E. Occurrence, temporal evolution and risk assessment of pharmaceutically active compounds in Doñana Park (Spain). J Hazard Mater. 2010; 183: 602- 608.
- Magiera S, Baranowska I, Kusa J. Development and validation of UHPLC-ESI-MS/MS method for the determination of selected cardiovascular drugs, polyphenols and their metabolites in human urine. Talanta. 2012; 89: 47-56.
- García-Galán MJ, Petrovic M, Rodríguez-Mozaz S, Barceló D. Multiresidue trace analysis of pharmaceuticals, their human metabolites and transformation products by fully automated on- line solid-phase extraction-liquid chromatography-tandem mass spectrometry. Talanta. 2016; 158: 330-341.
- Vazquez-Roig P, Kasprzyk-Hordern B, Blasco C, Picó Y. Stereoisomeric profiling of drugs of abuse and pharmaceuticals in wastewaters of Valencia (Spain). Sci Total Environ. 2014; 494-495: 49-57.
- Papageorgiou M, Kosma C, Lambropoulou D. Seasonal occurrence, removal, mass loading and environmental risk assessment of 55 pharmaceuticals and personal care products in a municipal wastewater treatment plant in Central Greece. Sci Total Environ. 2016; 543: 547-569.
- Ofrydopoulou A, Nannou C, Evgenidou E, Christodoulou A, Lambropoulou D. Assessment of a wide array of organic micropollutants of emerging concern in wastewater treatment plants in Greece: Occurrence, removals, mass loading and potential risks. Science of The Total Environment. 2022; 802: 149860.
- Kasprzyk-Hordern B, Dinsdale RM, Guwy AJ. The removal of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs during wastewater treatment and its impact on the quality of receiving waters. Water Res. 2009; 43: 363-380.
- Leusch FDL, Neale PA, Arnal C, Aneck-Hahn NH, Balaguer P, Bruchet A, et al. Analysis of endocrine activity in drinking water, surface water and treated wastewater from six countries. Water Res. 2018; 139: 10-18.
- Bendz D, Paxéus NA, Ginn TR, Loge FJ. Occurrence and fate of pharmaceutically active compounds in the environment, a case study: Höje River in Sweden. J Hazard Mater. 2005; 122: 195-204.
- Morasch B, Bonvin F, Reiser H, Grandjean D, de Alencastro LF, Perazzolo C, Chèvre N, Kohn T. Occurrence and fate of micropollutants in the Vidy Bay of Lake Geneva, Switzerland. Part II: micropollutant removal between wastewater and raw drinking water. Environ Toxicol Chem. 2010; 29: 1658-1668.
- Aydin E, Talinli I. Analysis, occurrence and fate of commonly used pharmaceuticals and hormones in the Buyukcekmece Watershed, Turkey. Chemosphere. 2013; 90: 2004-2012.
- Batt AL, Kostich MS, Lazorchak JM. Analysis of ecologically relevant pharmaceuticals in wastewater and surface water using selective solid-phase extraction and UPLC-MS/MS. Anal Chem. 2008; 80: 5021- 5030.
- Kostich MS, Batt AL, Lazorchak JM. Concentrations of prioritized pharmaceuticals in effluents from 50 large wastewater treatment plants in the US and implications for risk estimation. Environ Pollut. 2014; 184: 354-359.
- Cantwell MG, Katz DR, Sullivan JC, Ho K, Burgess RM. Temporal and spatial behavior of pharmaceuticals in Narragansett Bay, Rhode Island, United States. Environ Toxicol Chem. 2017; 36: 1846-1855.
- Cantwell MG, Katz DR, Sullivan JC, Shapley D, Lipscomb J, Epstein J, et al. Spatial patterns of pharmaceuticals and wastewater tracers in the Hudson River Estuary. Water Research. 2018; 137: 335-343.
- Oliveira TS, Murphy M, Mendola N, Wong V, Carlson D, Waring L. Characterization of Pharmaceuticals and Personal Care products in hospital effluent and waste water influent/effluent by direct- injection LC-MS-MS. Sci Total Environ. 2015; 518-519: 459-478.
- Schwartz H, Marushka L, Chan HM, Batal M, Sadik T, Ing A, et al. Pharmaceuticals in source waters of 95 First Nations in Canada. Can J Public Health. 2021; 112: 133-153.
- Nödler K, Voutsa D, Licha T. Polar organic micropollutants in the coastal environment of different marine systems. Marine Pollution Bulletin. 2014; 85: 50-59.
- Archer E, Petrie B, Kasprzyk-Hordern B, Wolfaardt GM. The fate of pharmaceuticals and personal care products (PPCPs), endocrine disrupting contaminants (EDCs), metabolites and illicit drugs in a WWTW and environmental waters. Chemosphere 2017; 174: 437-446.
- Osunmakinde CS. Tshabalala OS, Dube S, Nindi MM. Verification and validation of analytical methods for testing the levels of PPHCPs (Pharmaceutical & personal Health Care Products) in treated drinking water and sewage: Report to the Water Research Commission. 2013.
- Fries E, Mahjoub O, Mahjoub B, Berrehouc A, Lions J, Bahadir M.Occurrence of Contaminants of Emerging Concern (CEC) in conventional and non- conventional water resources in tunisia. Fresenius Environmental Bulletin. 2016; 25: 24.
- Moslah B, Hapeshi E, Jrad A, Fatta-Kassinos D, Hedhili A. Pharmaceuticals and illicit drugs in wastewater samples in north- eastern Tunisia. Environ Sci Pollut Res Int. 2018; 25: 18226-18241.
- Afsa S, Hamden K, Lara Martin PA, Mansour HB. Occurrence of 40 pharmaceutically active compounds in hospital and urban wastewaters and their contribution to Mahdia coastal seawater contamination. Environ Sci Pollut Res Int. 2020; 27: 1941-1955.
- Larsson DG, de Pedro C, Paxeus N. Effluent from drug manufactures contains extremely high levels of pharmaceuticals. J Hazard Mater. 2007; 148: 751-755.
- Singh V, Suthar S. Occurrence, seasonal variations, and ecological risk of pharmaceuticals and personal care products in River Ganges at two holy cities of India. Chemosphere. 2021; 268: 129331.
- Ashfaq M, Li Y, Rehman MSU, Zubair M, Mustafa G, Nazar MF, et al. Occurrence, spatial variation and risk assessment of pharmaceuticals and personal care products in urban wastewater, canal surface water, and their sediments: A case study of Lahore, Pakistan. Sci Total Environ. 2019; 688: 653-663.
- Ben W, Zhu B, Yuan X, Zhang Y, Yang M, Qiang Z, Occurrence, removal and risk of organic micropollutants in wastewater treatment plants across China: Comparison of wastewater treatment processes. Water Research. 2018; 130: 38-46.
- Duan L, Zhang Y, Wang B, Cagnetta G, Deng S, Huang J, et al. Characteristics of pharmaceutically active compounds in surface water in Beijing, China: Occurrence, spatial distribution and biennial variation from 2013 to 2017. Environmental Pollution. 2020; 264: 114753.
- An W, Duan L, Zhang Y, Zhou Y, Wang B, Yu G. Pollution characterization of pharmaceutically active compounds (PhACs) in the northwest of Tai Lake Basin, China: Occurrence, temporal changes, riverine flux and risk assessment. Journal of Hazardous Materials. 2022; 422: 126889.
- Behera SK, Kim HW, Oh JE, Park HS. Occurrence and removal of antibiotics, hormones and several other pharmaceuticals in wastewater treatment plants of the largest industrial city of Korea. Sci Total Environ. 2011; 409: 4351-4360.
- Lin AYC , Yu TH, Lateef SK. Removal of pharmaceuticals in secondary wastewater treatment processes in Taiwan. Journal of Hazardous Materials. 2009; 167: 1163-1169.
- Gaw S, Thomas KV, Hutchinson TH. Sources, impacts and trends of pharmaceuticals in the marine and coastal environment. Philos Trans R Soc Lond B Biol Sci. 2014; 369: 20130572.
- Klosterhaus SL, Grace R, Hamilton MC, Yee D. Method validation and reconnaissance of pharmaceuticals, personal care products, and alkylphenols in surface waters, sediments, and mussels in an urban estuary. Environ Int. 2013; 54: 92-99.
- Dodder NG, Maruya KA, Lee Ferguson P, Grace R, Klosterhaus S, La Guardia MJ, et al. Occurrence of contaminants of emerging concern in mussels (Mytilus spp.) along the California coast and the influence of land use, storm water discharge, and treated wastewater effluent. Mar Pollut Bull. 2014; 81: 340-346.
- Ali AM, Rønning HT, Sydnes LK, Alarif WM, Kallenborn R, Al-Lihaibi SS, Detection of PPCPs in marine organisms from contaminated coastal waters of the Saudi Red Sea. Science of The Total Environment 2018; 621: 654-662.
- Delahaut P, Brasseur PY, Dubois M. Multiresidue method for the detection of tranquillisers, xylazine, and a beta-blocker in animal production by liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2004 Oct 29; 1054: 373-378.
- Abou-Elwafa Abdallah M, Nguyen KH, Ebele AJ, Atia NN, Ali HRH, Harrad S. A single run, rapid polarity switching method for determination of 30 pharmaceuticals and personal care products in waste water using Q-Exactive Orbitrap high resolution accurate mass spectrometry. J Chromatogr A. 2019; 1588: 68-76.
- Bühring KU, Garbe A. Determination of the new beta-blocker bisoprolol and of metoprolol, atenolol and propranolol in plasma and urine by high-performance liquid chromatography. J Chromatogr. 1986; 382: 215-224.
- Ebele AJ, Oluseyi T, Drage DS, Harrad S, Abou-Elwafa Abdallah M. Occurrence, seasonal variation and human exposure to pharmaceuticals and personal care products in surface water, groundwater and drinking water in Lagos State, Nigeria. Emerging Contaminants. 2020; 6: 124-132.
- Fick J, Söderström H, Lindberg RH, Phan C, Tysklind M, Larsson DGJ. Contamination of surface, ground, and drinking water from pharmaceutical production. Environ Toxicol Chem. 2009; 28: 2522-2527.
- Giebu?towicz J, Stankiewicz A, Wroczy?ski P, Na??cz-Jawecki G. Occurrence of cardiovascular drugs in the sewage-impacted vistula river and in tap water in the warsaw region (Poland). Environmental Science and Pollution Research 2016; 23: 24337-24349.
- Ruan Y, Lin H, Zhang X, Wu R, Zhang K, Leung KMY et al. Enantiomer- specific bioaccumulation and distribution of chiral pharmaceuticals in a subtropical marine food web. Journal of Hazardous Materials. 2020; 394: 122589.
- Silva RA, Wang CC, Fernández LP, Masi AN, Flow injection spectrofluorimetric determination of carvedilol mediated by micelles. Talanta. 2008; 76: 166-171.









































































































































{kind=link}