The Functions of CHIP in Age Related Disease
- 1. Institute of Genetics and Molecular Medicine, University of Edinburgh, UK
Abstract
CHIP is a key component of the protein homeostasis or ‘Proteostasis’ network that maintains protein structure and function as a way to ensure the integrity of the proteome in individual cells and the health of the whole organism. Proteostasis influences the biogenesis, folding, trafficking and degradation of proteins. Originally identified as a Hsc70 associated protein and a co-chaperone CHIP has E3-ubiquitin ligase activity and also displays an intrinsic chaperoning ability. It has become clear that CHIP is a multi-functional protein with roles in cellular processes that go beyond
its co-chaperone activity. Not surprisingly, by unravelling the functions of CHIP, we are beginning to appreciate that loss of CHIP’s integrity can lead to the development of several serious pathological conditions. Here we will describe the key features of CHIPs structure and functions with an emphasis on the non-canonical activities of CHIP before concentrating on the role it plays in protecting against the age associated pathologies of neurodegeneration and cancer.
Keywords
• CHIP
• E3-ligase
• Chaperone
• Structure function
• Neurodegeneration
• Cancer
Citation
Ball KL, Ning J, Nita E, Dias C (2016) The Functions of CHIP in Age Related Disease. JSM Enzymol Protein Sci 1(1): 1006
ABBREVIATIONS
AD: Alzheimer’s Disease; CHIP: C-Terminus Of Hsc70 Interacting Protein; Hsc70: Heat Shock Cognate Protein 70; Hsp: Heat Shock Protein; RING: Really Interesting New Gene; Lys: Lysine; TPR: Tetratricopeptide Repeat; Cyp40: Cyclophilin 40; Hop: Hsp70/Hsp90 Organizing Protein; PP5: Protein Phosphatase 5; HX-MS: Hydrogen-Deuterium Exchange Mass Spectrometry: Ppiase: Peptidylprolyl Isomerase; Bag-1: BCL2 Associated Athanogene 1; IRF-1: Interferon Regulatory Factor-1; CFTR: Cystic Fibrosis Transmembrane Conductance Regulator: AMP: Adenosine Monophosphate: AMPK: Adenosine Monophosphate Kinase; HSF1: Heat Shock Factor Protein 1; α-Syn: α-Synuclein: Enos: Endothelial Nitric Oxide Synthase; TLR: Toll-Like Receptor; IFN β: Interferon β; PKC ζ: Protein Kinase Cζ: Sirt6: Sirtuin-6; LKB1: Liver Kinase B1; HIF1A: Hypoxia-Inducible Factor 1-Alpha; CMA: Chaperone-Mediated Autophagy; CASA: Chaperone Assisted Selective Autophagy; MEFS: Mouse Embryonic Fibroblasts; UPS: Ubiquitin Proteasome System; APP: Amyloid Precursor Protein; Aβ: Amyloid β; PD: Parkinson’s Disease; EGFR: Epidermal Growth Factor Receptor; Erbb2: Erb-B2 Receptor Tyrosine Kinase 2; NF-κB nuclear Factor Kappa-Light-Chain-Enhancer Of Activated B Cells; ER: Estrogen Receptor; GR: Glucocorticoid Receptor; PRMT5: Protein Arginine Methyltransferase 5; E2F1:E2F Transcription Factor 1; TG2: Transglutaminase 2; Bcl-2: B-Cell Lymphoma 2; TRAF2: TNF Receptor-Associated Factor 2; EMT: Epitheliaap1lMesenchymal Transition; MMP9 :Matrix Metallopeptidase 9; MMP2 Matrix Metallopeptidase 2; TGF-β: Transforming Growth Factor Beta; AP1: Activator Protein 1
INTRODUCTION
Protein homeostasis or ‘Proteostasis’ is the term used to encompass the elaborate systems involved in maintaining protein structure and function as a way to ensure the integrity of the proteome in individual cells and the health of the whole organism. Proteostasis involves a highly complex network of processes that influence the biogenesis, folding, trafficking and degradation of proteins. The dual function E3-ubiquitin ligase and molecular chaperone CHIP (C-terminus of Hsc70 interacting protein) was originally identified as a link between protein folding and protein degradation. In addition to being a co-chaperone that targets partially-folded or unfolded client proteins for proteasomal degradation dependent on its E3-ligase activity, CHIP can also function independently of its Hsp-partners to control the structure and function of proteins through its intrinsic chaperone and/or E3-ligase activities. Besides its functions in ubiquitindependent proteasome-mediated degradation, CHIP is implicated in chaperone dependent autophagy and protein trafficking. Not surprisingly, by unravelling the functions of CHIP, we are beginning to appreciate that loss of CHIP’s integrity can lead to the development of several serious pathological conditions. Here we will introduce the general functions of CHIP with an emphasis on its non-canonical activities, followed by the role of CHIP in the age related process of neurodegeneration and tumorigenesis.
Structure of chip
CHIP contains a conserved C-terminal U-box domain and N-terminal TPR domain, which are separated by a coiled-coil region [1]. Structurally, the U-box is like a RING-finger except that it lacks metal chelating residues, playing a similar role to the RINGfinger domain in targeting the substrate and mediating ubiquitinconjugation to it [2]. RING-finger type E3 ligases were originally considered to be ‘passive’ i.e. they do not contain a catalytic site but instead function as scaffolds by placing the substrate and ubiquitin-charged E2 into close proximity [2]. However, recent crystallography data on RING domains in complex with E2 and ubiquitin partners suggests a more active role in determining E2~Ub conformation [3,4]. CHIP can ubiquitinate substrates to form either Lys48-linked chains [5] or Lys63-linked chains [6], dependent on the E2-partner [7]. Whilst Lys48-linked substrates can be degraded by the proteasome and Lys63-linked chain modification results in a variety of outcomes [8,9]. The crystal structure (PDB ID: 2C2V) for ‘nearly’ full-length murine CHIP (aa25-304) was solved in 2005, and showed the C-terminus of CHIP in complex with Ubc13-Uev1a, an E2 heterodimer that can catalyze the formation of Lys63-linked ubiquitin chains. In this structure a hydrophobic ridge in Ubc13 binds into a U-box hydrophobic groove [10]. A subsequent structure (PDB ID: 2OXQ) for the CHIP U-box from Danio rerio in complex with UbcH5a has also been solved and shows a similar architecture to the murine structure [11,12].
The TPR-domain of CHIP (residues 26-131 in human) is located at the N-terminus. A TPR-domain is a 34 amino acid motif which forms a pair of anti-parallel helices that has been identified in a broad range of proteins [13]. It is assembled in tandem arrays of 3-16 motifs [14] and the distribution of these arrays can be found separated from each other or arranged together [15]. Generally speaking, the TPR motif can be described as a mediator of protein-protein interactions that facilitates the assembly of protein complexes [15]. In CHIP, three TPR domain units generate six helices that consist of three pairs of antiparallel helices. The hydrophobic surface of the TPR domain is responsible for the interaction with the carboxy-terminus of Hsc/ p70 or Hsp90 [1,16]. In 2005, the crystal structure (PDB ID: 2C2L) of murine CHIP in complex with a Hsp90 C-terminal decapeptide was published. It showed that the TPR domain of CHIP is similar to TPR domains in other proteins, e.g. HOP/Cyp40/PP5 etc. In this structure, the Hsp90 peptide was bound in the TPR domain hydrophobic pocket, which forms a distinct two-carboxylate clamp. The electrostatic interaction is contributed primarily by an aspartic acid from Hsp90 and lysine residues in the TPR domain of CHIP [10]. A recent structure of CHIP and Hsp70 lid-tail (HsHsc70541-646Δ626-638) revealed that residues in addition to the Hsp70 C-terminal peptide ‘PTIEEVD’, namely the α-helical lid sub-domain, interact with CHIP in order for Hsp70 to act as a CHIP substrate and post-translational modifications of the lid can regulate Hsp70-CHIP binding affinity [17].
The coiled-coil region of CHIP exists as a linker between the TPR- and U-box domains. It is essential for dimer formation which in turn is required for CHIP to be fully active as an E3 ligase [18]. It is of note that, in the murine structure, CHIP forms an asymmetric dimer with different conformations in the coiledcoil region for each protomer. In one subunit, a hairpin is formed by two straight helices. In the other subunit, one of the helices is broken into two separated α-helices. When the structures for CHIP-Hsp90 peptide (2C2L) and CHIP- Ubc13-Uev1a (2C2V), are overlaid the TPR domain in the dimer will block access to one of the U-box binding sited for the E2, thus only one U-box can bind with E2 [10] and is presumable sufficient for catalysis.
Recently, using HX-MS, it was shown that the apo-CHIP TPR domain is very flexible [19]. When bound to a Hsp70/90 C-terminal peptide the TPR-domain becomes more stable and organized. By using a mutation at Lys30 of the TPR of CHIP, the entire TPR domain can be ‘locked’ (Figure 1).
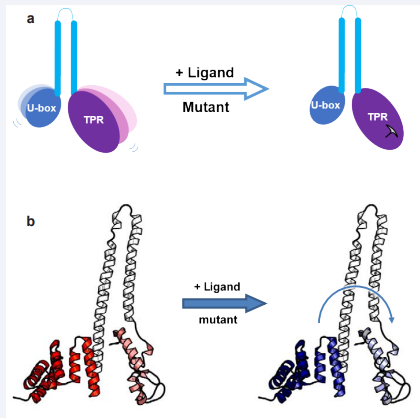
Figure 1: Inter-domain allosteric regulation of CHIP. The allostery signal transmits from the TPR domain of CHIP to the U-box domain. The TPR domain of CHIP is very flexible. By binding to Hsp70, or by introducing a mutation in the TPR domain, the TPR domain becomes ‘locked’ and allosterically reduces the flexibility of U-box. (a) A cartoon showing that locking of the TPR domain makes the U-box domain more stable. (b) A structure showing the conformational inhibition signal transfers from the N-terminal TPR domain to C-terminal U-box. Red means flexible. Blue indicates stable.
Also, changes to the flexibility of the TPR domain affect U-box conformation [20]. The E3-ligase activity of CHIP decreased significantly upon Hsp70/90 peptide binding or with the replacement of Lys30 with an Ala; this indicates that an intra-molecular allostery can be transmitted from the TPR domain to the U-box domain (Figure 1). Similar results were reported for Cyp40 bound to the Hsp peptides ‘XXXMEEVD’ with evidence presented that the peptide can inhibit the PPIase activity of Cyp40 [21]. Finally, the data on CHIP allosteric regulation suggests that cellular experiments using a K30A mutant of CHIP must be interpreted with care [20].
Chip functions
Canonical activity of chip as a co-chaperone: CHIP was discovered by the Patterson group [1] as a binding protein for Hsc70; it was enriched in muscle tissue and could act as a negative regulator of Hsp70/Hsp40 chaperone function in vitro [1]. Subsequently, CHIP was shown to interact with Hsp90, as well as Hsc/Hsp70, and could induce ubiquitination of the glucocorticoid receptor leading to its degradation through the ubiquitin-proteasome pathway [16]. Thus, CHIP modulated protein triage decisions impacting on the balance between folding and degradation of chaperone client proteins. The role of CHIP in protein triage was further defined when it was demonstrated to have U-Box dependent E3-ubiquitin ligase activity [22]. This led to the definition of CHIP as a co-chaperone, which together with BAG-1 [6,23], could shift the activity of the Hsc/Hsp70 chaperones from a protein folding- to a protein degradation-machine [22- 24]. Thus, the early days of CHIP research focused on its role as an E3-ligase that ubiquitinated unfolded or mis-folded proteins targeting them for ubiquitin dependent proteasome degradation only in the presence of its chaperone partners [23-25]. This canonical CHIP pathway has been covered in detail in a number of recent reviews [26-28].
Non-canonical functions of chip: The first hint that the functions of CHIP may be more complex than simply a constitutive ubiquitinating co-factor for the core molecular chaperones, came from Meacham et al., (2001) [28]. This study showed that CHIP has substrate specificity and did not catalyse the degradation of all Hsp70-dependent clients. Thus, whereas CHIP over expression led to the ubiquitination and degradation of the Hsp70 client protein CFTR it had no effect on a range of other Hsp70 substrates (including the transferin receptor and luciferase [29,30]). Furthermore, in contrast to the ability of CHIP to inhibit Hsp70 mediated folding in vitro [1,16], over-expression of CHIP in cells could increase the folding capacity of Hsp70 in an E3-ligase independent manner [30]. This lead to the suggestion that the association of CHIP with Hsp70 did not always convert this class of chaperone from a folding to a degradative mode [30]. In agreement with this we have shown that CHIP over expression or depletion differentially affects the levels of the interferon regulated transcription factor IRF-1 dependent on the cellular conditions. Thus, in cells that are recovering from heat stress CHIP is a negative regulator of IRF-1 steady state levels whereas in cycling cells CHIP enhances IRF-1 protein levels [31].
We now know that CHIP has several non-canonical functions which either do not require chaperone partners, which can be negatively regulated by CHIP association with Hsp’s or where CHIP and Hsp70 cooperate to regulate processes other than the degradation of unfolded or mutant proteins through the proteasome. Some examples of this are given below:
Chip chaperone activity: Although CHIP was originally identified as a co-chaperone for Hsc/Hsp70, it later became apparent that CHIP also had its own intrinsic chaperone activity. Thus, over expression of CHIP in fibroblasts, was able to increase the refolding of proteins in cells following thermal denaturation [30]. Under these conditions inhibition of Hsp70 abolished the effects of CHIP on protein folding suggesting that CHIP might be functioning to enhance Hsp70-dependent chaperone activity. Furthermore, enhancement of Hsp70 ‘folding’ function was independent of CHIP E3-ligase activity. However, subsequent studies went on to show that CHIP could bind to and refold heat-denatured luciferase in vitro and in cells, independent of Hsp70. We can therefore conclude that CHIP has an intrinsic chaperone activity that is elevated during heat stress [32] and that it may also function to activate, or cooperation with, Hsp70 in a substrate folding pathway [30]. CHIP can physically interact with native p53 leading to its ubiquitination [20]. In addition, binding of CHIP to an N-terminal domain is reported to prevented p53 from irreversible thermal inactivation [33]. In this study CHIP was found to bind preferentially to the mutant conformation of p53 and to restore the DNA binding activity of p53 following heat inactivation. The authors proposed that CHIP might therefore be a direct chaperone for chromatin bound p53 helping it to maintain a transcriptionally active conformation under stress conditions [33]. Perhaps the best evidence for the physiological role of CHIP as a chaperone comes from studies on the AMP activated kinase (AMPK). In the case of the AMPK, CHIP can regulate the catalytic α-subunit by promoting LKB-1-mediated phosphorylation, which in turn leads to an increase in its specific activity. CHIP binding to AMPK causes a conformational change in the α-subunit suggesting that CHIP can chaperone native as well as non-native proteins to affect changes in their activity [34].
Chip as an activator of hsf1-mediated transcription: In response to heat stress a portion of total CHIP protein moves from the cytoplasm into the nucleus where it can be found in complex with active HSF1, a transcription factor that regulates heat shock protein expression [35,36], at heat shock elements [37]. Thus, whereas over expression of CHIP leads to trimerization and activation of HSF1, knock-down of CHIP decreases the activity of HSF1 and these changes occur independently of any change in the half-life of the transcription factor [37,38]. Although the mechanism by CHIP promotes the activity of HSF1 remains unclear CHIP and HSF1 are reported to form a direct interaction under conditions of heat stress [39]. Fascinatingly CHIP appears to cause the re-localisation of HSF1 [37]. The interaction between CHIP and HSF1 can occur independently of Hsp70, and indeed there is evidence that CHIP might recruit Hsp70 to the HSF1 transcription complex under some conditions [37]. In this situation however, Hsp70 would oppose rather that support CHIP function leading to inhibition of HSF1 [40].
Docking dependent substrate ubiquitination by chip: As described above the canonical pathway for CHIP is that it ubiquitinates unfolded or mutant substrates in collaboration with Hsp70 and targets them for degradation. In this pathway CHIP does not interact directly with the substrate but substrate recognition is mediated by Hsp70. However, there is also evidence that CHIP can ubiquitinate substrates independently of Hsp70 and that in this pathway modification requires the direct binding of CHIP to its substrate. We have shown that CHIP binds directly to IRF-1 [31] to carry out docking-mediates site specific ubiquitination of the IRF-1 DNA-binding domain [41]. In this scenario conditions which block access to the E3-binding site, such as when IRF-1 is in its DNA bound conformation [41], inhibit substrate ubiquitination. Interestingly, when IRF-1 is subject to unfolding during mild heat stress binding of CHIP is inhibited and it becomes a poor substrate for CHIP E3-ligase activity (Landre, Mativa, Ning and Ball, unpublished observation). However, whether Hsp70 would restore ubiquitination under these conditions remains to be seen. Docking-dependent ubiquitination of native IRF-1 and, a second substrate p53, are inhibited by Hsp70 as binding leads to inhibition of U-box activity. Thus binding of full-length Hsp70, or Hsp70 mimetic peptides, to the TRP-domain of CHIP can act as allosteric regulators that affect CHIP substrate-docking and U-box activities [20]. This study is supported by the observation that CHIP-dependent monoubiquitination of Smad1/5 [42] is inhibited by Hsp70 and by data using α−synuclein where suppression of mono-ubiquitination by BAG-5 is Hsp70-mediated [43]. As IRF-1, like HSF1 (see above), is a transcriptional factor it is interesting to speculate that the relocalisation of CHIP to the nucleus under stress conditions may have wider implications for transcriptional regulation than the control of Hsp expression [37].
Role for chip in regulation of protein trafficking: As referred to above over expression of CHIP can lead to a redistribution of HSF1 suggesting that CHIP may play roles in protein trafficking. For example, CHIP has been shown to interact with and ubiquitinate Daxx, a death domain associated protein and transcriptional suppressor, to counter its proapoptotic activity. The ubiquitination of Daxx does not signal degradation but temporarily sequesters it into an insoluble fraction from which it can later be released when the cells are in the recovery phase [44,45]. Similarly, CHIP does not target eNOS for ubiquitination and proteasome-dependent degradation but partitions soluble eNOS into an insoluble and inactive cellular compartment [46]. However, unlike Daxx, eNOS regulation by CHIP occurs in unstressed cells, suggesting that CHIP may play a more general role in protein trafficking [46].
Regulation of protein activity by chip: One of the areas to receive recent attention, in terms of CHIP activity, is innate and adaptive immunity. CHIP plays a role in antigen aggregation and presentation during maturation of antigen presenting cells as part of the co-chaperone network [47]. Perhaps more surprisingly, CHIP can coordinate the assembly of active Toll-Like Receptor signalling complexes, specifically for TLR4 and TLR9, leading to the activation of downstream signal components and the production of antiviral cytokines including IFNβ [48]. The authors of this study suggest that as well as acting as scaffolding protein for active TLR complex assembly, CHIP may be involved in the recruitment of Src and PKCζ to TLR4/9 and their subsequent activation. How CHIP activates these signalling kinases is unclear, however evidence is presented that the K63-ubiquitin ligase activity of CHIP may be required [10,48]. Another example of CHIP playing a more direct role in regulating protein activity is SirT6. SirT6 is a lysine deacetylase/ADP ribosylase involved in DNA repair and metabolism. In this case CHIP maintains SirT6 activity by preventing its degradation. This requires CHIP E3- ligase activity and the authors speculate that CHIP-dependent ubiquitination of SirT6 at K170, prevents other E3-ligases from K48-linked polyubiquitnation and degradation of SirT6 [49].
Autophagy and chip: In addition to the well-studied link between CHIP and proteasome mediated degradation there is growing evidence that CHIP can also take part in autophagy. As the link between autophagy and age related diseases are well established [50], autophagy regulation could be import for the role of CHIP in neuroprotection (see below). Thus, whilst a decrease in CHIP protein levels in neuronal cells induces autophagosome formation degradation of the classic autophagy marker protein p62 is decreased [51] leading the authors to speculate on the role of CHIP in autophagic flux. Studies in CHIP null mice indicate a role for CHIP in mitophagy and the maintenance of energy metabolism in the CNS [52]. The functions of CHIP spread beyond canonical autophagy, thus it plays a role in antigen presentation as a component of the ubiquitin-dependent chaperone-assisted selective autophagy (CASA) process [47,53]. Further, CHIP takes part in a second form of chaperone-dependent autophagy named Chaperone-Mediated Autophagy (CMA) [54,55]. An example of CHIPs role in CMA is the hypoxia-inducible factor and transcription factor HIF1A. In the case of HIF1A, CHIP is required for its ubiquitination by K63 linked ubiquitin, which is an early step in CMA-dependent degradation. However, the requirement for CHIP activity in CMA is likely to be substrate specific [56,57].
CHIP in age related disease: Perhaps one of the most striking phenotypes of surviving CHIP null mice is the effect on longevity [58]. The CHIP null-mice have a 60% decrease in lifespan compared to wild-type animals and this is accompanied by increased anatomical ageing. These mice have signs of increased oxidative stress in the brain [58] and, are hypersensitive to DNA-damaging agents [49]. Ageing and cellular senescence are enhanced by various stress conditions, including oxidative stress, defects in proteasome-mediated degradation and DNAdamage [59]. Likewise, CHIP expression is dynamic and is activated during cellular senescence (i.e. ageing) in normal cells to facilitate targeting of proteins to the proteasome, including the degradation of p53, a key modulator of senescence. However, when CHIP is silenced (using siRNA) in human embryonic lung fibroblasts (HFL-1), hallmarks of premature senescence are seen: irreversible cell growth arrest, induction of p16, and accumulation of β-gal positive cells [60]. This could be explained in part by the role of CHIP in preventing oxidative stress; CHIP knockdown results in pronounced accumulation of oxidized proteins and the addition of antioxidants only slightly delayed senescence [59,60]. Oxidative stress is likely to result from an impaired ubiquitin-proteasome system (UPS), which in turn promotes oxidative stress, triggering a vicious cycle. Mouse embryonic fibroblasts (MEFs) derived from CHIP knock-out mice (CHIP-/-) show increases longevity of polyubiquitin- and proteasomalsubstrates [60] as well as in-creases in oxidized protein and lipids [58]. Conversely, over expressing CHIP has cytoprotective effect due to CHIP-mediated degradation of oxidized proteins by the proteasome preventing imbalances in protein quality control and reduced longevity [60]. Further, CHIP binds to, and regulates, SirT6 and this interaction defines an intersection between proteostasis and the epigenetic regulation in aging [49]. In addition to the effects of CHIP on longevity and cellular aging there is also growing evidence that CHIP can protect form aging related diseases such as neurodegeneration and cancer.
Neurodegeneration
General neuroprotective function(s) of chip linked to oxidative stress: There is evidence that the neuroprotective effect of CHIP can be linked, at least in part, to its ability to protect neurons and CNS cells from oxidative stress [52,58,60]. In turn the antioxidant effects of CHIP appear to be linked to its ability to ubiquitinate a flavoenzyme, NAD (P) H: Quinone Oxidoredctase 1 (NQO1); an enzyme that is induced in response to electrophilic and/or oxidative stress [61] and interacts with the 20S proteasome to prevent the degradation of some proteins [62]. Whilst the level of CHIP is down-regulated with ageing NQO1 shows a corresponding increase [61]. CHIP also interacts with endonuclease G (EndoG), a mitochondrial endonuclease responsible for mediating caspase-independent DNA fragmentation which leads to cell death following acute oxidative stress [63,64]. Cells depleted of CHIP have an increased susceptibility to cell death following treatment with the oxidant H2 O2 (~1.7 fold higher) and increased levels of EndoG both before and after treatment. Under normal conditions, EndoG interacts with Hsp70 and is ubiquitinated by CHIP in a Hsp-dependent manner. Treatment with the oxidizing agent H2 O2 abolishes the interaction with Hsp70, and in turn with the TPR domain of CHIP, and EndoG translocates to the nucleus for DNA fragmentation. Observed age-dependent decreases in CHIP levels most likely explain the corresponding increase in EndoG and enhanced susceptibility to oxidative stress.
Chip in alzheimer’s disease: AD is one of the most common forms of late-onset dementia, characterized by memory loss, language disturbances and executive dysfunction as well as a decline in cognitive function. Pathological hallmarks of the disease are intracellular neurofibrillary tangles (NFT) composed of filamentous hyperphosphorylated tau and senile plaques composed of extracellular deposits of amyloid-β (Aβ). Accumulation of these abnormal protein aggregates is associated with neuronal loss. There is growing evidence for the importance of the molecular chaperons and ubiquitin ligases in the fate of hyperphosphorylated tau and the toxic form of the amyloid precursor protein (APP).
CHIP appears to have an important role in APP metabolism. APP undergoes ubiquitination on its C-terminal cytoplasmic region [65]. Stable CHIP-APP interactions are detected in different cell cultures and post-mortem human brain. Evidence of this is the increase in the levels of normal APP after CHIP over expression and proteasome inhibition. Pull-down experiments have shown an increase in the levels of ubiquitinated APP interacting with CHIP. APP undergoes proteolytic cleavage by β- and α- secretases to form C99 and C83 amino acid C-terminal APP fragments (CTFs), respectively. In cells over expressing CHIP CTFs [66] accumulate and are more stable. In addition, CHIP might also influence the accumulation of the β- amyloid peptide (Aβ) product of γ- secretase action on C99, accelerating Aβ removal and protecting against accompanying toxicity. In conclusion, on one hand CHIP interacts with HSPs to stabilize normal APP and on the other it assists in the ubiquitination of a subpopulation of APP molecules that are destined for proteasome degradation [66].
An important characteristic of AD is the formation NFTs that are composed of the microtubule-associated protein tau. During the development of disease pathology tau becomes hyperphosphorylated, detaches from the axonal microtubules and aggregates [67]. Tau lesions in post-mortem human tissue are immune-positive for CHIP which interacts and specifically ubiquitinates tau in vitro and in vivo [68]. CHIP decreases the amount of total tau in a TPR-domain dependent manner [69] suggesting a role for CHIP chaperone activity or Hsp binding. CHIP deficient mice have reduced Hsp70 mRNA and protein level while the level of tau mRNA is unchanged suggesting that the substantial increase in phospho-tau and total-tau is due to role of CHIP in tau protein degradation [70]. Thus, in the absence of CHIP there is an increased accumulation of ubiquitin-negative, phosphorylated tau species in the presynaptic compartment. In addition, these mice exhibit increased caspase-3, cleaved caspase-3 and caspase-3-cleaved tau. However, neither hyperphospholylation nor caspase-3 cleaved tau are sufficient to induce aggregation of tau in the absence of CHIP, suggesting that ubiquitination has an important role in the formation of the tau aggregates [70].
In cell cultures, CHIP increases the insoluble tau fraction composed of high molecular weight aggregated tau species, whist expression of Hsp70 reduces it [68]. Moreover, phosphotau is an Hsp90 substrate and it facilitates its dephosphorylation and refolding preventing degradation. Thus inhibition of the Hsp90 refolding pathway and over expression of CHIP result in the degradation of phosphorylated tau [71]. In AD patient brains, there is an inverse relationship between CHIP and PHFtau (paired helical filaments) accumulation [72]. Finally, over expression of full length tau or a tau isoform results in changes in mitochondrial distribution a phenomenon that can be rescued by CHIP [69,73].
Chip in parkinson’s disease: PD is a progressive neurodegenerative disease caused by selective degeneration of dopaminergic neurons. The pathological hallmark of the disorder is the presence of intracytoplasmic fibrillary aggregates called Lewy Bodies (LB) the main component of which is misfolded, aggregated α-Syn. Under certain cellular conditions α-Syn starts to form oligomers, which ultimately leads to aggregation into amyloid-like fibrils [74-76]. Thus, the role of proteasome degradation and protein chaperone machinery in regulated α-Syn levels and conformation is of major interest in PD research.
In cell culture models and in human brain tissues, α-Syn, co-localizes with CHIP and Hsp70 in intracellular inclusions. Over expression of CHIP results in a reduction in the number of cells containing α-Syn inclusions [77]. Moreover, CHIP causes morphological changes transforming large inclusions into micro-aggregates dispersed throughout the cytoplasm in a TPRdependent manner [77]. It is reported that the degradation of α-Syn can be achieved both by the proteasome pathway in a TPR dependent manner and by U-box domain-mediated lysosomal degradation [74]. It is not surprising therefore that CHIP overexpression reduces the levels of intermediate, toxic and high molecular weight α-Syn oligomers and rescues the resultant cytotoxicity [78].
In vitro and in vivo ubiquitination assays confirmed the ability of CHIP to mono-, multi mono- and poly-ubiquitinate α−Syn [79]. The E3 ligase activity of CHIP, and thus α-Syn ubiquitination, can be inhibited by BAG5, which causes enhanced dopamine neuron death in a model of PD [43,79]. BAG5, CHIP and α-Syn exist within the same complex and their interaction is dependent on the Hsp70 chaperone. Finally, α-Syn co-immuno precipitates with Hsp70 and Hsp90 [80], and inhibition of Hsp90 resulting in decreased α-Syn oligomerization and toxicity in Drosophila, yeast and cellular models of PD [81-83], suggesting that α-Syn is a Hsp90 substrate which could be ubiquitinated and degraded by CHIP in an Hsp70 dependent manner. However, we should perhaps introduce a note of caution as much of the research on CHIP in relation to α-Syn has used cell systems in which either mutant forms of CHIP or tagged forms of α-Syn are over expressed. Therefore, it is still not clear whether direct CHIP-dependent ubiquitination of endogenous α-Syn in healthy individuals or in human pathologies is physiologically relevant. What is clear is that CHIP plays a central role in maintaining healthy aging in the brain and CNS [52,58].
Cancer
Accumulating evidence indicates that CHIP predominantly functions as a tumour suppressor, preventing cell proliferation and/or promoting apoptosis in breast, gastric, prostrate, pancreatic and colorectal cancers, glioma and hepatoma [84,85]. These cellular effects are the result of CHIP-mediated regulation of oncogenic proteins, including the estrogen receptor ERα, ErbB2, androgen receptor, histone deacetylase 6, hypoxiainducible factor-1A (HIF-1a), C-Myc, p65 and EGFR, which in turn influences NF-kβ signaling, the Akt pathway and the epithelialmesenchymal transition [85-87] (Figure 2).
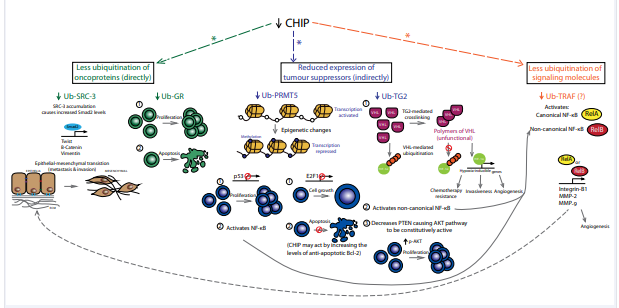
Figure 2: Changes in CHIP activity reduce the ubiquitination of oncoproteins, tumour suppressors and signalling molecules. The net cellular effects include overall suppression of apoptosis and enhanced cell proliferation, cell growth (including anchorage-independent growth), angiogenesis, chemotherapy resistance, epithelialmesenchymal transition (thus promoting metastasis and invasiveness). Interactions between signalling pathways are meant to illustrate protein-protein interactions downstream of CHIP. They do not necessarily mean that all processes occur in a specific cancer. *E3 ligase activity that is TPR domain-dependent. Abbreviations: CHIP (C-Terminus of Hsc70 Interacting Protein); Ub (Ubiquitinated); SRC-3 (Steroid Receptor Co-Activator 3); GR (Glucocorticoid Receptor); PRMT5 (Protein Arginine Methyltransferase 5); TG2 (Transglutaminase 2); VHL (Von Hippel-Lindau); HIF-1a (Hypoxia-Inducible Factor 1a); P (Phosphorylated); NF-Kb (Nuclear Factor Kappa Beta) And MMP (Matrix Metalloproteinase’s)
Different groups have tried to understand the mechanism underlying the low CHIP gene expression detected across tumours. Two cancer gene databases (The Cancer Genome Atlas and COSMIC) revealed no “loss-of-function” mutations. However, analyses of copy-number variations detected loss of the gene in 17.5% of renal and 45% of ovarian cancer patient samples.
Alternatively, epigenetics is a plausible explanation for the low CHIP mRNA levels. Gastric, colorectal and breast cancer patients have significant hyper-DNA-methylation in the CHIP promoter [84,88]. Although CHIP levels did not correlate with promoter methylation and it was hypothesized that other epigenetic changes, such as histone deacetylation, may account for this discrepancy [89]. Thus the mechanism causing reduced CHIP expression in tumours is controversial, and the mechanism may differ between cancer type and from patient to patient.
Effects of chip on cell growth parameters: Typical of a tumour suppressor protein, CHIP over expression inhibits oncogenic signaling pathways, cell migration and anchorage independent growth and induces cell death. On the other hand reduced levels of the CHIP protein promote tumour formation metastasis and this is thought to be mediated primarily by the degradation of oncogenic proteins [85]. An example of this is CHIP’s effect on glucocorticoid receptor (GR) levels, GR is expressed in 50-70% of invasive breast cancers and can even be a biological and prognostic biomarker of lower grade, ER-positive luminal breast cancer. GR has anti-proliferative and anti-apoptotic functions and its levels decrease during cancer progression [90]. In situ, CHIP ubiquitinates GR in a dose-dependent manner and transfecting an E3 ligase-defective CHIP shows dominantnegative effects. Importantly, CHIP co-immunoprecipitates with the HC8 particle of the S5a proteasome subunit and such direct interaction most likely occurs in the context of the assembled 26S proteasome complexes.
It may seem counterintuitive in the context of cancer that tumours show lower levels of CHIP compared to surrounding healthy tissue. As discussed above, cells depleted of CHIP become prematurely senescent, possibly due to defects in redox and protein homeostasis [59,60], which could prevent cancer growth [91]. However, CHIP causes degradation of strategic oncoproteins, such as protein arginine methyltransferase 5 [85]. PRMT5 causes epigenetic changes that promote tumourgenicity and is upregulated during the development and progression of several cancers including lymphoma, leukemia and breast, gastric, colorectal and ovarian cancers [92-94]. This type II methyltransferase functions as an oncoprotein by methylating histone 4 at arginine 3 and/or histone H3 at arginine 8 to silence the expression of tumour suppressors, including p53 and RB [87,94]. Alternatively, PRMT5 can methylate non-histone substrates to activate signalling molecules such as E2F1, p53, RelA/p65, epidermal growth factor receptor (EGFR), RAD9, and programmed cell death 4 [87,93]. Methylation of E2F1 reduces its ability to suppress cell growth and to promote apoptosis, rending cancer cells with a survival advantage. Methylation of p65 activates the NF-κβ and TNF signalling pathways [87]. CHIP mediated the K48-linked polyubiquitination of PRMT5, targeting it for proteasomal degradation. Such post-translational modification was dependent on both the TPR and U-box domains of CHIP for PRMT5 recognition and ubiquitination, respectively. Hsp90 inhibitors decreased PRMT5 protein expression in a dose-dependent manner, and this was dependent on CHIP expression. Again, this suggests that the degradation of PRMT5 may be regulated by the molecular chaperone system involving CHIP, Hsp90 and Hsp70. Interesting, over expression of CHIP under these conditions increased cell death and inhibited cell growth; such effects may be mediated by CHIP-dependent down-regulation of PRMT5 expression [87].
CHIP expression is negatively correlated with transglutaminase 2 (TG2) in renal cell carcinoma. The catalytic core of TG2 interacts with the TPR domain of CHIP, followed by its K48-linked ubiquitination and degradation. CHIP increases the steady-state levels and stability of TG2. Pharmacological inhibition of Hsp70 inhibits CHIP-mediated TG2 degradation and conversely, over-expression of Hsp70 enhances its degradation [85]. Therefore, TG2 and PRMT5 degradation seem to be regulated by CHIP in the canonical pathway where it cooperates with the core molecular chaperones. The increased expression of TG2 may be specific to renal cancer, since expression is greatly decreased in breast cancer and glioma possibly due to hypermethylation [85]. In renal cancer, TG2 degradation might be hindered during tumour development possibly due to alterations in the balance between CHIP and TG2 expressions, conditions that favour cell growth, migration, metastasis and angiogenesis [85].
Effects of chip on cell signalling in cancer: Alterations to CHIP expression lead to imbalances in protein homeostasis, affecting multiple signalling pathways both directly and indirectly. When over-expression CHIP in a human gastric cancer cell line the expression level of Akt was unchanged, although there was a reduction in its phosphorylated, active form (p-AKT at either Ser473 or Thr308), leading to reduced cellular proliferation. Additionally, CHIP over-expression markedly decreased the expression of the anti-apoptotic gene Bcl-2, causing increased apoptosis [95]. Therefore, in both gastric and breast cancer cells, where CHIP expression is low, Bcl-2 and Akt are upregulated, leading to anchorage-independent cell growth [87,88]. In breast cancer, SRC-3-mediated upregulation of Smad2 (a signal transducer in the TFG-β signalling pathway) was also observed. The latter is related to aggressive phenotypes in tumours by directly upregulating twist, β-catenin and vimentin, which promote EMT and thus allowing benign tumours to metastasize. Such invasive and metastatic character was abolished when SRC3 was silenced [87,88]. The expression of RelA/p65 and RelB (involved in the canonical and non-canonical NF-κβ pathways, respectively) were also upregulated. Driving this dysregulation was the decrease in CHIP-mediated suppression of TRAF2 expression (possibly by ubiquitination and degradation), a positive regulator of both branches of the NF-κβsignalling. NFκβ and AP-1 are transcription factors that bind to the promoter of the gelatinases (matrix metalloproteinase) MMP-2 and MMP9 and of the integrin-β-1 gene, regulating their activity. These are closely associated with cancer invasion: MMP-2 promotes cleavage of the extracellular matrix while MMP-9 modulates permeability of the vascular endothelium [95]. High expression of integrin-β-1 has been detected during invasion, angiogenesis and metastasis of breast cancer and glioblastoma. In line with this, CHIP over expression inhibited the activity of the NF-κβ pathway and caused decreased levels of MMP-2, MMP-9 and integrin-β-1, leading to diminished migration and invasion of gastric cancer cells [95].
DISCUSSION AND CONCLUSION
As we discover more about the dual functions of CHIP as an E3 ubiquitin-ligase and chaperone working as part of the molecular chaperone network, or more independently, as a regulator of native protein function, it is clear that CHIP occupies a central position in proteostasis. As the loss or impairment of CHIP function(s) has the potential to cause pronounced homeostatic defects and disease pathologies, it has been put forward as a potential therapeutic target in a range of conditions [86,96,97]. Yet despite growing knowledge on the biology of CHIP we still know relatively little about how its structure is related to its functions and activities. For example, how are the intrinsic chaperone and E3-ligase activities of CHIP coordinated or differentially regulated to ensure that the endogenous protein is able to respond in multiple proteostatic pathways. Although the use of mutated exogenous proteins expressed in cells can take us some way down the path to understanding the activities and regulation of CHIP, it has its limitations as illustrated by the effects of mutations and interacting proteins on CHIPs activity though changes in structural flexibility linked to allosteric regulation [19,20]. Moving forward, engineering tools that isolate either the E3-ligase or chaperone activity of endogenous CHIP would allow us to inhibit or activate its functions independently. Such tools would also likely have applications in the field of therapeutics.
REFERENCES
46. Jiang J, Cyr D, Babbitt RW, Sessa WC, Patterson C. Chaperone-dependent regulation of endothelial nitric-oxide synthase intracellular trafficking by the co-chaperone/ubiquitin ligase CHIP. J Biol Chem. 2003; 278: 49332-49341.
86. Paul I, Ghosh MK. A CHIPotle in physiology and disease. Int J Biochem Cell Biol. 2015; 58: 37-52.
91. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013; 75: 685-705.








































































































































