Emerging CRISPR Technologies Available Through Resource Sharing
- 1. Addgene Inc., USA
Abstract
CRISPR is a popular approach for genome engineering, as it requires very few components (Cas9 and a gRNA) and can be adapted to knockdown, knockout or activate expression of specific genes in a wide variety of organisms. Although CRISPR is certainly the most straightforward genome engineering technique in use today, there are still significant technical barriers to using CRISPR. The requirement for a Protospacer Adjacent Motif (PAM) sequence adjacent to a putative target sequence reduces the number of genomic loci that can be targeted by Cas9. Furthermore, the ability of Cas9 to cleave genomic loci with less than perfect homology to the gRNA means that “off-targets” can be unintentionally modified. Finally, the need to build new gRNA expression plasmids for each DNA target limits the speed with which researchers can carry out genome engineering experiments. Recently, several CRISPR reagents have been developed to circumvent these limitations and increase the speed and efficiency with which CRISPR can be used for genome engineering. The purpose of this review is to highlight novel CRISPR technologies and discuss how reagent sharing contributes to the rapid expansion of CRISPR research.
Keywords
• Genome Engineering
• CRISPR
• Cas9
Citation
McDade JR, Waxmonsky NC (2016) Emerging CRISPR Technologies Available Through Resource Sharing. JSM Genet Genomics 3(1): 1008.
ABBREVIATIONS
CRISPR: Clustered Regularly Interspersed Short Palindromic Repeats; Cas9: Cas-Associated Endonuclease 9; gRNA: guide RNA; HDR: Homology-Directed Repair; NHEJ: Non-Homologous End Joining
BACKGROUND AND INTRODUCTION
CRISPR/Cas9 is a simple, yet powerful genome engineering system capable of modifying the genome in a wide variety of organisms and cell types. CRISPR consists of two requisite components, 1) a non-specific endonuclease, Cas9 and 2) a ~75 nucleotide “guide” RNA (gRNA) composed of a “scaffold sequence” necessary for Cas9 binding and a user-defined ~20 nucleotide “targeting sequence” which determines the genomic DNA target. When expressed simultaneously, the gRNA and Cas9 form a complex that is capable of binding and generating a double-strand break (DSB) in target DNA. Cas9-induced DSBs are typically repaired through the error-prone Non Homologous End Joining (NHEJ) repair pathway and the end resulting in a functional “knockout” of the target gene [1–3]. CRISPR has since been adapted to generate specific genome edits using Homology Directed Repair (HDR) and activate or repress target genes[4–6]. CRISPR is certainly one of the most versatile systems for genome engineering, but it is not without limitation. First, the number of suitable target sites is limited by the necessity for a Protospacer Adjacent Motif (PAM) downstream of the genetic loci to be modified [7]. Also, Cas9 is capable of cleaving genomic loci with less than perfect homology to the gRNA targeting sequence (termed “off-targets”) in addition to the desired target sequence [8]. Finally, although CRISPR is much faster and easier to use than previous genome editing techniques, researchers are still required to identify suitable target sequences and develop gRNA and Cas9 expressing plasmids for use in experiments. This review covers recent advances in CRISPR technology that circumvent these limitations, with a focus on reagents that are currently available to academic researchers through Addgene1 and discusses how reagent sharing accelerates CRISPR research.
Emerging Trends in CRISPR
Increasing the Reach of CRISPR with Novel PAM Binding Variants of SpCas9 Wild-type S. pyogenes Cas9 (heretofore referred to as SpCas9) is the most widely used CRISPR endonuclease for genome engineering experiments. SpCas9 can cleave virtually any genomic sequence provided the target is unique to the rest of the genome and located immediately upstream of a PAM sequence (5’NGG’3 for SpCas9). The SpCas9 PAM sequence is ubiquitous throughout the genome, but not every genetic locus will have a suitable target-PAM sequence due to the absence of a PAM or an abundance of potential off-targets for a given target sequence. In such a case, the genetic loci in question cannot be modified using CRISPR. To circumvent this limitation, the Joung lab at Massachusetts General Hospital (MGH) used a bacterial mutagenesis screen to identify mutants of SpCas9 that were capable of binding and cleaving target sequences upstream of non-NGG PAM sequences. The result of this work was the identification and characterization of three novel SpCas9 mutants that bind non-NGG PAM sequences: SpCas9 VRER (binds NGCG PAM; Addgene #65773), SpCas9 EQR (binds NGAG PAM; Addgene #65772) and SpCas9 VQR (binds NGAN or NGNG PAM; Addgene #65771). Importantly, SpCas9 PAM variants have been demonstrated to cleave endogenous target sites with similar cutting efficiency and specificity when compared to wild-type Cas9 [9]. Theoretically, inclusion of these the SpCas9 variants into the CRISPR arsenal doubles the number of PAM sites in the human genome and will facilitate modification of genetic loci that were previously refractory to CRISPR modification [9].
Increasing the Specificity of CRISPR with “Enhanced” specificity SpCas9s
CRISPR/Cas9 can be used to make precise genome modifications at genomic loci by designing gRNA targeting sequences with perfect homology to the DNA target. However, Cas9 is also able to modify genomic loci with less than perfect homology to the gRNA targeting sequence (e.g. “off-targets”) [8]. The specificity of the CRISPR system refers to the ability of the Cas9-gRNA complex to discriminate between the desired target site and all of the potential off-targets within the genome. Proper gRNA design can compensate for much of the inherent off-target cleavage activity of Cas9 and increase efficiency of on-target cleavage, but variants of Cas9 that are inherently more specific may prove useful as researchers and clinicians look toward using CRISPR in the clinic [10]. Previous efforts to increase CRISPR specificity utilized SpCas9 with a single mutated cleavage site (“SpCas9-Nickase” or “Cas9n”) or a complete cleavage incompetent Cas9 (“dCas9”) fused to the non-specific end nuclease FokI [11–13]. Cas9n and dCas9-FokI increase specificity by restricting DSBs to regions of the DNA that have two properly targeted Cas9 molecules bound in close proximity. Off-target binding, on the other hand, results in an easily reparable DNA nick (for Cas9n) or no cleavage at all (for dCas9-FokI). A variety of Cas9n and dCas9-FokI reagents are available for distribution through Addgene.
Unfortunately, the major strength of these approaches is also the major limitation, as not every genomic locus will have two suitable target-PAM sequences in close enough proximity to generate a DSB. Structural biology studies have shed light on how the Cas9-gRNA complex binds and cleaves target (and off-target) DNA [7,14], and this information has been used to rationally design Cas9 variants with higher specificity than wild type SpCas9. These “high specificity” Cas9s use the same basic gRNA design principles as wild-type SpCas9 but are able to cleave target sequences with far greater specificity than wild-type SpCas9. Importantly, high specificity Cas9s require a single gRNA, thus circumventing the requirement for multiple suitable PAM Target sites within a target gene. Two different high specificity Cas9s are available through Addgene: eSpCas9 from the Zhang lab at the Broad Institute of MIT and Harvard (Addgene #71814) and SpCas9-HF variants from the Joung lab at MGH (https:// www.addgene.org/browse/article/16457/) [15,16].
Increasing the Speed of gRNA Design and Cloning with Empty gRNA backbones and validated gRNAs
Despite the abundance of ready-to-use Cas9-expressing plasmids available to researchers, the necessity to design and clone gRNA-expressing plasmids remains a significant bottleneck when carrying out CRISPR experiments. Guide RNAs can be designed manually by searching the genomic sequence of target genes for unique 18-20 nucleotide sequences upstream of a PAM site and comparing each potential target to the rest of the genome to identify potential “off-targets”. Although “off targets” are readily apparent, “on-target efficacy” is much more difficult to predict when gRNAs are manually designed. Luckily, several gRNA design programs are now freely available, which are capable of identifying potential Target-PAM sequences and ranking the gRNA targeting sequences based on predicted “on target” and “off-target” efficacy [10,17].
Once the desired gRNA targeting sequences have been designed, they must be synthesized and cloned into the appropriate expression plasmid (if using a plasmid-based expression system). Researchers commonly create “empty” gRNA plasmids, which contain an RNA-Pol III promoter to drive expression of the gRNA and the 3’ gRNA scaffold sequence required for Cas9 binding, which facilitate construction of the desired gRNA-expression plasmid in a single cloning step. Addgene currently has approximately 250 “empty gRNA plasmids” optimized for expression in cell types and organisms ranging from bacteria to humans. Empty gRNA-expression plasmids are often accompanied by a detailed cloning protocol which serves as a step-by-step guide on how to design and clone targeting oligos into the vector. Alternatively, researchers can avoid constructing gRNA-expression plasmids altogether by using a pre-constructed gRNA-expression plasmid containing a “validated gRNA”. Validated gRNA plasmids are gRNA-expression plasmids that have been demonstrated to be efficacious (e.g. published in a peer-reviewed journal) and thus, represent a “ready-to-use” gRNA expression plasmid for CRISPR experiments. Currently, about 350 “validated gRNA” plasmids are available from Addgene.
Resource sharing accelerates CRISPR research
Almost 1,000 CRISPR plasmids have been deposited to Addgene by more than 100 research labs since 2012 (Figure 1).
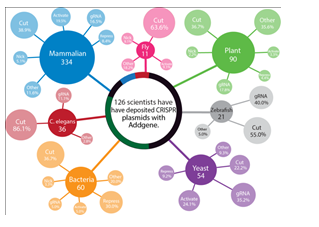
Figure 1: Roughly 1000 CRISPR plasmids are available to researchers at academic and nonprofit research institutions through Addgene. The above figure represents a snapshot of the plasmids that are available for expression in specific model organisms, further sub-divided by specific CRISPR application. The CRISPR collection at Addgene facilitates genome engineering in variety of model organisms including microbes, yeast, worms, plants and mammals and spans the most common CRISPR applications including gene knockout (using wild-type Cas9 “cut” or Cas9-nickase), gene activation and repression. Additional uses of CRISPR that are represented in the collection (labeled “other” above) include DNA purification, reporter/tag knock-in and visualization of specific genomic loci using fluorescently labeled Cas9. The gRNA label refers to both empty gRNA vectors and validated gRNA plasmids.
The collection spans most of the major CRISPR applications including knockout and activation or repression of target genes. Since the first CRISPR plasmids were made available through Addgene in 2012, Addgene has distributed >54,000 CRISPR plasmids to ~15,000 scientists across the globe. The availability of “ready-to-use” reagents removes the unnecessary burden of having to develop CRISPR reagents de novo for every experiment and provides researchers access to the most recent cutting edge CRISPR technologies. Furthermore, the availability of detailed protocols for CRISPR plasmids makes any necessary cloning much easier for researchers. Overall, resource sharing increases the speed with which experiments can be performed, which is highlighted by the number of publications citing CRISPR plasmids from the Addgene repository–Addgene CRISPR plasmids have been cited ~600 times. Addgene plays an active role in procuring cutting edge CRISPR reagents, and is currently looking to expand the CRISPR repertoire to include both pooled and arrayed gRNA libraries and expand upon plant and microbial CRISPR reagents. Given the rapid expansion of the CRISPR field, reagent sharing will undoubtedly play a critical role in making new and novel technologies available to the broader research community to facilitate genome engineering research.
1 Addgene is a nonprofit plasmid repository located in Cambridge, Massachu setts USA. Researchers deposit plasmids with Addgene and Addgene archives and distribute plasmids to academic and nonprofit institutions world-wide
ACKNOWLEDGEMENTS
The authors thank all of the laboratories that have deposited CRISPR plasmids with Addgene. The authors thank Joanne Kamens for her critical reading of this manuscript.
Conflict of Interest
The authors are members of Addgene, the nonprofit plasmid repository.









































































































































