Sexually Dimorphic Phenotype of Pulmonary Artery Hypertension as a Function Epigenetic Regulation of Soluble Epoxide Hydrolase
- 1. Department of Physiology, New York Medical College, USA
- 0. Authors made equal contributions
Abstract
Increases in pulmonary epoxyeicosatrienoic acids (EETs), caused by inhibition of soluble epoxide hydrolase (sEH, encoded by the Ephx2 gene), promote pulmonary artery hypertension (PAH). We hypothesized that the estrogen-dependent methylation and hypoxamir-driven mechanisms work synergistically to downregulate the pulmonary Ephx2 gene expression and promote PAH in a sex specific manner. We used male and female Sugen-hypoxia (SuHx) rats implanted with a pulmonary artery (PA), radio-telemetry transducer catheter to continuously record changes in PA pressure during of hypoxia exposure for three weeks. In the early stages of PAH (hypoxia for 8 days), both sexes of SuHx rats developed significant elevations of PA pressure, responses that were in a female-intensified manner. Treatment of SuHx rats with TPPU (sEH inhibitor), significantly augmented elevations of PA pressure, while overexpression of sEH via administration of adenovirus encoded the Ephx2 gene mitigated the elevation. This suggests that EETs promote PAH. Meanwhile, female SuHx rats consistently displayed increases in DNA methylation of the Ephx2 gene, followed by the downregulation of sEH and increases in pulmonary EETs. In the later stage of PAH (hypoxia for 21days), the female predominant sex difference in incremental elevations of PA pressure disappeared. This disappearance is attributed to the initiation of microRNA-411-3p (a hypoxamir)-driven downregulation of sEH, concomitant with the upregulation of CYP2C23 (pulmonary EET synthase) in male rats. In conclusion, both female-specific estrogen-dependent methylation and male-predominant hypoxamir-driven silencing of the Ephx2 gene are responsible for the EET-mediated potentiation of PAH in a time specific manner.
Keywords
Sex; Soluble epoxide hydrolase; Pulmonary artery hypertension; DNA methylation; Hypoxamirs
Citation
Kandhi S, BenRahoma G, Ishiko S, Yang YM, Wolin MS, et al. (2022) Sexually Dimorphic Phenotype of Pulmonary Artery Hypertension as a Function Epigenetic Regulation of Soluble Epoxide Hydrolase. JSM Sexual Med 6(2): 1084.
INTRODUCTION
The differential expression of genes that are either selective targets of, or regulated by sex hormones, contribute significantly to the sex-different phenotype in the cardiovascular system and pulmonary circulation. In this regard, the pulmonary artery hypertension (PAH) is a classical paradigm with the consensus that females bear high susceptibility to PAH development [1,2]. Germline mutation-mediated changes in estrogen signaling seem to be responsible for the female susceptibility of PAH, as indicated by clinical correlations between the polymorphism of gene(s) involving estrogen metabolism and high occurrence of PAH in women [1,3-5]. Particularly, autosomal BMPR2 (bone morphogenetic protein receptor type II) mutation turns out to be one of the most important genetically based alterations for the female susceptibility to IPAH [6,7]. However, only recently has the female predisposition to PAH as a function of epoxyeicosatrienoic acids (EETs; metabolites of arachidonic acids from cytochrome P450), attracted our attention. This is not only because EETs are pulmonary vasoconstrictors [8-11], but more importantly because soluble epoxide hydrolase (sEH encoded by the Ephx2 gene; responsible for the degradation of EETs to their bio-inactive diols-DHETs) can be transcriptionally regulated by sex hormones and pathological circumstances of pulmonary circulation [10,12-15]. Indeed, the Ephx2 gene is modulated by estrogens via the DNA methylation mechanism [15]. This epigenetic event incurs the phenotype of female-facilitated, EETmediated cardiovascular outcomes [3,8,16-20].
Epigenetics is defined as the interaction of genes with their surrounding environment, leading to a specific phenotypic expression [21], which is not the result from changes in genomic nucleotide sequences, but rather evoked by modifications in DNA regulatory molecules, such as altered small noncoding RNAs (micro-/or miRNAs), or through chemical modification of the DNA via methylation of a gene promoter [22]. As such, DNA methylation of the Ephx2 gene to intensify EET-induced pulmonary vasoconstrictions forms a molecular basis that supports EETs as instigators of PAH. Alternatively, as a primary pathological characteristic of PAH, hypoxia serves as a master regulator of specific miRNA(s), termed hypoxamir(s), to modify downstream target gene expressions [23]. In this regard, the significance of hypoxamir-triggered modulation of the pulmonary Ephx2 gene has been left unaddressed.
To date, there are few studies that clarify the connection between the estrogen downregulation of the Ephx2 gene and female predisposition to PAH. Nor does evidence indicate whether, and if so how, hypoxia modulates the Ephx2 gene expression during the process of PAH. To this end, we hypothesized an epigenetic-based regulation of the pulmonary Ephx2 gene during PAH development, by which, both estrogendependent methylation and hypoxamir-driven (independent of estrogen) pathways work in concert to conduct the sexually dimorphic phenotype of PAH. We used radiotelemetry with transducer catheters implanted in rat pulmonary arteries (PAs) in order to dynamically monitor changes in PA pressure as a function of sex and EETs.
MATERIALS AND METHODS
Animals
Twelve-week-old male (M) and female (F) Sprague-Dawley (SD) rats purchased from Charles River Laboratories were used. The Sugen-hypoxia model of pulmonary artery hypertension (SuHx-PH) that exists with replicable characteristics of human PAH was created by subcutaneous injection of Sugen 5416 (20mg/kg), followed by exposure of rats to hypoxic cages. Hypoxia was induced by continuous infusion of a gas mixture of 50% air and 50% nitrogen to generate 10% oxygen. Each airtight cage containing only one rat, was equipped with its own radio-telemetry receiver and drinking water bottle that could be changed without affecting the hypoxic atmosphere. In separate groups, male SuHx rats were receiving TPPU (sEH inhibitor; 0.5 mg/150 ml/day) in their drinking during their exposure to hypoxia, while some females had received intratracheal instillation of adenovirus (5x109 pfu/rat) encoding the Ephx2 gene (Ad-sEH) before their treatment with SuHx. Changes in PA pressure were recorded continuously for up to 21 days.
All protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conform to the guidelines of the National Institutes of Health and the American Physiological Society for the use and care of laboratory animals.
Adenovirus transfection
Rats were anesthetized with Ketamine/Xylazine (70/6 mg/ kg, ip) and kept at 600 supine head up position to receive 0.3ml of intratracheal instillation of adenovirus (5x109 pfu/rat) using a rat model PennCentury microsprayer. The rat Ephx2 gene was cloned into pEnter-C-GFP vector. The high-tittered and purified adenovirus encoding the Ephx2 gene (Ad-sEH) was purchased from Vigene Bioscience. Control GFP adenovirus (CV10001), was used as transfection controls. The specificity and efficiency of the intratracheal transfection of Ad-sEH have been verified by our preliminary studies.
Radio-telemetry measurement of PA pressures
Rats were implanted with the pressure transducer (TA11PA-C40) for PA pressure recording. We used the transdiaphragm approach of surgery to implant the transducer, as described [24]. Briefly, rats were anesthetized with Ketamine/ Xylazine (70/6 mg/kg, ip) and then ventilated through tracheal intubation. After laparotomy, a midline incision in the diaphragm was made to expose the heart. Right ventricle (RV) was punctured, and the catheter was carefully inserted into the RV and further advanced into the PA. The live pressure waveform trace (shown in Figure 2a) confirmed the correct position of the catheter in PA. The catheter was then fixed on the RV wall. The diaphragm was closed, and the full expansion of the lungs was restored. The transmitter was implanted in the peritoneal cavity, fixed to the abdominal wall, then followed by closing the abdomen and skin. After the operation, rats were maintained in separate cages for ~2 weeks. They were then injected with Sugen and transferred to the hypoxic cages. After that, a continuous recording of in vivo changes in PA pressure was setup in a cycle of ten-second recording period every ten-minute for 8 and 21 days, respectively. The pressure pulse and digital data (e.g. systolic, diastolic, heart rate, and other parameters) were saved periodically.
RV catherization for measurement of the systolic pressure (RVSP)
The measurement was performed in separate groups of female rats after they had received intratracheal instillation of the Ad-sEH and were exposed to SuHx for 7, 14 and 21 days, respectively. The procedure was described in detail previously [12]. Briefly, rats were anesthetized with inhalation of Isothesia (isoflurane). A pressure catheter was placed in the right jugular vein and advanced into the RV for measuring RVSP. The pressure catheter (Transonic Systems Inc, Ithaca, NY) was equipped with a pressure transducer (TRN050; Kent Scientific, Torrington, CT) and transducer amplifier (Kent Scientific). Changes in RVSP were recorded on PowerLab (ADInstruments, Colorado Springs, CO) and analyzed with Chart V8 software.
DNA methylation of the Ephx2 gene
Procedures for DNA/CG methylation of the promoter of Ephx2 gene were described in detail previously [15]. Briefly, PAs were freshly isolated from male and female rats before (normoxia) and after their exposure to SuHx for 21 days. Based on rat Ephx2 promoter sequence, methylation-specific PCR (MSP) on isolated PAs was performed after bisulfite modification of genome DNA by bisulfite genomic sequencing (BGS). Genomic DNA was extracted from isolated PAs and MSP primers were designed by using MethPrimer program (https://www.urogene.org/methprimer).
LC/MS/MS-based measurements of lung EETs and DHETs
At the end of experiments, lungs were excised for EET measurements. As described in detail [14,20], esterified EETs and DHETs from tissue extracts were quantified via LC/MS/MS (AB ScieX, Qtrap 3200). Protein concentration of samples determined by the Bradford method (Bio-Rad, Hercules, CA), was used to normalize the detected lipids that were expressed as picograms per microgram protein. Total EETs and DHETs were the sum of all four isoforms of EET and DHET.
Quantitative real time RT-PCR
Total RNA was extracted from isolated pulmonary tissues using TRIzol solution (Sigma-Aldrich), and treated with RNasefree DNase. Total RNA (2 μg) from each sample was subjected to real-time quantitative PCR (LightCycler, Roche Diagnostics, Indianapolis, IN), according to the manufacturer’s instructions and a QuantiTect SYBR Green PCR kit (Qiagen). The primers were designed in-house and synthesized by Fisher Scientific customer services. Quantification was reported, using proprietary software, as cycle threshold (Ct). The sEH expression was normalized to GAPDH or actin. A relative quantitation method (ΔΔCt) was used to evaluate the expression of the gene in different groups of samples. All amplified products were verified on a 1.5% agarose gel.
Western blot analysis
Isolated rat lungs were pulverized in liquid N2. Equal amounts of total protein extracted (50 µg) were loaded onto and separated by 10% SDS-PAGE gel electrophoresis and transferred to a PVDF membrane. The membrane was probed with specific primary antibodies for sEH (62 Kd with 1:1000 dilution) or CYP2C23 (58 Kd with 1:1000 dilution), and appropriate secondary antibodies conjugated with horseradish peroxidase. The specific bands were visualized with a chemiluminescence kit (Thermo Scientific, Rockford, IL) and normalized to GAPDH (38 Kd with 1;2000 dilution). The X-ray film was scanned into a computer and band densitometry was digitalized with UN-SCAN-IT software (Silk Scientific, Orem, UT).
Chemicals
All chemicals, unless specified otherwise, were obtained from Sigma Chemical (St. Louis, MO). Primary antibodies for sEH and CYP2C23 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), while TPPU from Cayman Chemical Co. (Ann Arbor, MI).
Statistics
Data are presented as mean ± SEM, and n refers to the number of rats or blots. Statistical analyses were performed using GraphPad Prism 6 software. One- and two-way ANOVA followed by the Tukey-Kramer post hoc test were used to compare the difference among multiple groups. Student’s t-test was used to compare the difference between two groups. Statistical significance was accepted at a level of p< 0.05.
RESULTS
Time- and dose-dependent upregulation of pulmonary sEH, as a function of intratracheal transfection of Adshe
Figure 1 shows that the specific staining for sEH (DAB, brown color) at PA and pulmonary venules (PV) (b & c) was significantly increased, accompanied by stronger GFP staining (e & f) compared to controls (a & d). With consistency, Figure 1g shows a significant upregulation of sEH protein in PAs of the rats transfected with one dose (1x) of Ad-sEH for 7 and 14 days (7D &14D) respectively, a response that declined at 21D posttransfection, yet maintained in rats that received a second dose (2x) of Ad-sEH at 14D.
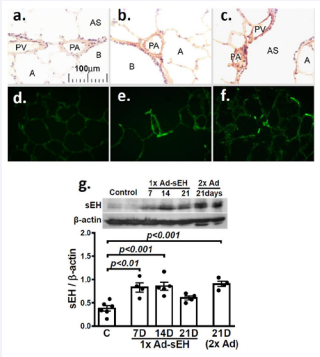
Figure 1: (a-c) Immunohistochemistry of sEH (brown color, DAB staining). (df) Fluorescence images of sEH-GFP in lung sections of female controls (a, b) and female rats transfected with intratracheal instillation of Ad-sEH for 7 days (b, c, e, f), showing a significantly increased sEH staining in Ad-sEH transfected rats (b & c), accompanied by stronger GFP staining (e & f) compared to controls (a & d). (g) shows pulmonary artery sEH expression in controls and in rats transfected with a single dose (1x) of Ad-sEH for 7, 14 and 21 days, and 2 doses of Ad-sEH (2x) for 21 days, indicating a upregulation of sEH protein in PAs of the rats transfected with one (1x) and two doses (2x) of Ad-sEH (n =3 blots with oneway ANOVA).
PA: pulmonary artery; PV: pulmonary venule; B: bronchiole; AS: alveolar sac; A: alveolus.
EET-mediation of female-intensified elevation of PA pressure (in vivo)
To study sex-different generation and severity of PAH, we continuously recorded changes in PA pressure in SuHx-PH rats implanted with a PA radio-telemetry transducer catheter. Original PA waveform tracing (Figure 2a) confirms the catheter position. Figure 2b shows that under a comparable basal PA pressure, rats treated with SuHx developed a time-dependent elevation of PA pressure, however, the incremental increases in female rats occurred earlier and in a greater magnitude compared to males, suggesting a female-favorable elevation of PA pressure. To evaluate roles of EETs in the female-favorable generation of PAH, male rats received TPPU to mimic the female pattern of reduced sEH activity. Indeed, TPPU-treated male SuHx rats exhibited a significantly greater elevation of PA pressure than untreated males, expressing a response pattern similar to that of females. Alternatively, female rats that had been transfected with Ad-sEH to overexpress pulmonary sEH displayed significantly lesser elevations of RVSP (indicator of PA pressure) than those without the transfection, a magnitude that became comparable to male SuHx rats (Figure 2c). These data confirm EETs as essential players in the female-facilitation of PAH.
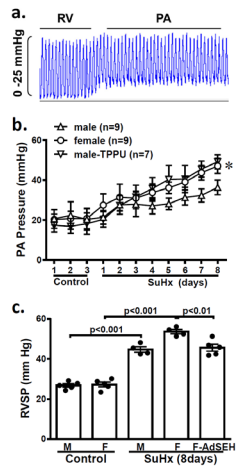
Figure 2: Radio telemetry recording of PA waveforms (a) and summarized mean PA pressure (b), and catheterization measurement of right ventricular systolic pressure (RVSP; c) before and during SuHx-treatment for 8 days in male, female and male rats treated with TPPU (b; n=7-9). Data show significantly greater elevation of PA pressure in SuHx female and TPPU-treated male rats than untreated SuHx male rats. Whereas SuHx female rats receiving Ad-sEH (c; n=4- 5) significantly attenuated their elevations of RVSP compared to those without the transfection. *Significant difference from male (two-way ANOVA).
Downregulation of pulmonary sEH increased tissue EETs in female SuHx rats (in vitro)
Figure 3 indicates that the physiological downregulation of sEH in the lungs of female rats was maintained after exposure to SuHx for 8 days (a), paralleled with increases in total tissue levels of EETs (c) and decreases in DHETs (d), leading to a greater ratio of EETs to EHDTs in females compared to their male littermates (e), even though their CYP2C23 (major EET synthase in the rat lung) expressions remains comparable at this time point (b). Thus, pulmonary EET levels are proportional to the magnitude of elevated PA pressure.
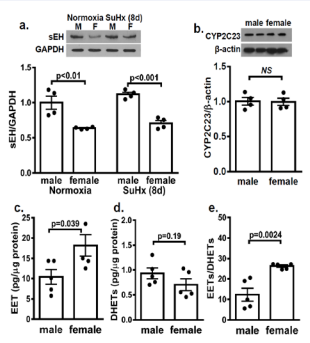
Figure 3: Female (F) downregulation of pulmonary expression of sEH (a; n=3 blots with two-way ANOVA) in comparison with males (M) was maintained in normoxia and after treated with SuHx for 8 days, while their CYP2C23 expressions (b; n=3 blots with one-way ANOVA) were comparable. As a result, female rats display increases in total tissue levels of EETs (c) and decreases in DHETs (d), leading to a greater ratio of EETs to EHDTs (e; n=5 with one-way ANOVA).
Disappearance of sex-differences in the incremental elevation of PA pressure after PAH was established
Intriguingly, as shown in Figure 4, extending SuHx treatment up to 21 days the incremental difference in the elevation of PA pressure between sexes disappeared, which inspired us to follow up with the changes in pulmonary EET metabolism. We found that unlike in the early stage of SuHx treatment (8 days), wherein suppressed pulmonary sEH expression existed only in females, associated with a comparable EET synthase in both sexes (Figure 3a & 3b). After exposure of rats to SuHx for 21 days, both male and female rats displayed downregulation of sEH (Figure 5a), while upregulation of CYP2C23 occurred only in males (Figure 5b). This male-specific upregulation of EET synthase and downregulation of EET hydrolase caused significant increases in pulmonary EETs in male SuHx rats, leading to a comparable EETs/ DHETs ratio in both sexes (Figure 5c), eliminating differences in their incremental elevations of PA pressure (Figure 4). Thus, the results reveal a specific time frame illustrating a sex-different downregulation of sEH during PAH progression and confirm further, the crucial roles of EETs in the control of PA pressure.
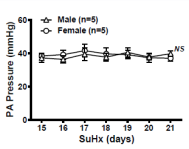
Figure 4: Radio telemetry recording of mean PA pressure showing comparable increases in PA between male and female rats during SuHx-treatment for 15-21 days. (n=5 for each with two-way ANOVA).
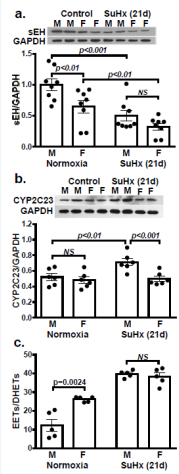
Figure 5: sEH and CYP2C23 protein expressions (a & b; n=3 blots for each) in lungs of male and female controls and SuHx (21 days) rats. Data show a downregulation of sEH in both sexes of rats after SuHx treatment for 21 days, while upregulation of CYP2C23 in males only, leading to a comparable EETs/ DHETs ratio (c; n=4-5) in SuHx-treated males and females. (Two-way ANOVA).
Female-favorable DNA methylation of the Ephx2 gene was unaffected by chronic hypoxia
Figure 6 shows that in physiological/normoxic conditions, female PAs expressed increases in DNA methylation in the promoter of their Ephx2 gene, leading to a greater ratio of methylated to unmethylated (M/U) DNA than male vessels (a & b). The consequential suppression of sEH mRNA in female PAs (Figure 6c), confirms the methylation-dependent transcriptional silencing of the Ephx2 gene. Interestingly, the pathological challenge with SuHx did not significantly affect the methylation status, as evidenced by Figure 7 showing a statistically equivalent M/U ratio before and after SuHx treatment. Thus, the next step was to identify an alternative pathway responsible for the downregulation of pulmonary sEH in male SuHx rats (Figure 5a).
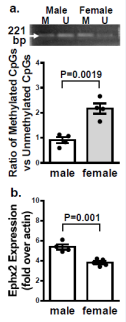
Figure 6" Original and summarized data (a) of the ratio of methylated (M) to unmethylated (U) Ephx2 DNA, and Ephx2 mRNA (b) in PAs isolated from normal male and female rats. Data show significant increases in the ratio of methylated to unmethylated Ephx2 DNA (a), associated with downregulation of Ephx2 mRNA expression in female rats (b). (n=3 for each group with one-way ANOVA).
Male-specific stimulation of miRNA-411-3p silenced the Ephx2 gene, as a function of chronic hypoxia
To test specific roles of hypoxamirs in the methylationindependent modification of sEH, we reviewed the data of small/micro RNA (miRNA) sequencing performed after hypoxic exposure for 21 days, and found an increase in pulmonary miR411-3p, a hypoxamir that can directly target the Ephx2 gene at the position in 977-984 of Ephx2 3’UTR with a high probability of preferential conservation to suppress its expression (http:// targetscan.org). To verify the result, quantitative RT-PCR for miR-411-3p and western blot analysis for its target protein (sEH), as well as sEH products (DHETs) were assessed. As shown in Figure 8, after 21 days of hypoxia, male SuHx rats exhibited an upregulation of pulmonary miR-411-3p mRNA, consequentially followed by a downregulation of sEH protein expression (Figure 5a), and increases in the ratio of EETs/DHETs (Figure 5c), while a statistically insignificant increase in miR-411-3p was observed in female SuHx rats (P=0.08). This novel finding indicates a miR-411-3p-driven modulation of Ephx2/sEH during PAH development.
DISCUSSION
Salient findings of the study are as follows. First, the use of radio telemetry with PA-located transducer catheter to dynamically record changes in PA pressure allows real-time monitoring of in vivo generation, severity, and progression of PAH in response to SuHx, as a function of sex (male vs female) and pulmonary EETs (down- vs over-expression of sEH) (Figures 1-2 & 4). Second, a direct correlation, if not a causal relationship, between EETs and PA pressure was revealed, as indicated by the results that the greater increases in PA pressure in female and TPPU-treated male SuHx rats were proportional to their pulmonary levels of EETs (Figure 3), while elimination of sex differences in the pulmonary EET levels were followed by comparable elevation of PA pressure between sexes. (Figure 4,5). Third, the estrogen-dependent methylation of PA Ephx2 gene to suppress sEH expression (Figure 6); this epigenetic event elicits the earlier and greater elevation of PA pressure in female SuHx rats (Figure 1 & 7). Finally, we demonstrated for the first time, that the male-specific downregulation of the Ephx2 gene (Figure 5a) was driven by chronic hypoxia-triggered activation of hypoxamir/miR-411-3p (Figure 8), which in combination with enhanced EET synthesis (Figure 5b), contributes to the delayed increases in pulmonary EETs and further augmented PA pressure in male SuHx-PH rats. Thus, the sexually dimorphic phenotype of PAH is primarily determined by changes in sEH expression and EET metabolisms within a specific time frame, wherein, estrogen-dependent methylation of the Ephx2 gene potentially intensifies EET-mediated elevations of PA pressure in the early stage of PAH. Alternatively, chronic hypoxia-evoked, hypoxamirinduced downregulation of Ephx2/sEH incurs the exacerbation of EET-mediated elevations of PA pressure during the later stage of PAH.
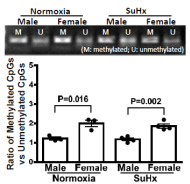
Figure 7: Original and summarized data of DNA methylation of the Ephx2 gene in PAs isolated from male and female rats before (normoxia) and after exposure to SuHx for 21 days, showing an similar NDA methylation pattern between and after SuHx treatment. (n=4 for each group with two-way ANOVA).
EET-dependent potentiation of PAH generation and severity, as a function of estrogen-induced methylation of the Ephx2 gene
PAH is a complex disease that initially provokes pulmonary vasoconstrictions, then chronically forms plexiform lesion to occlude PAs [25], followed by right heart hypertrophy, dysfunction, and failure [1,26, 27]. As with female-predominant prevalence [28], the incidence of PAH may reflect a genetic predisposition to the disease [3, 8], which in general, takes place under physiological or sub-physiological conditions. Thus, any pulmonary vasoconstrictors with intensified bioavailability by female hormones, such as EETs, can trigger PAH signaling in a female-susceptible manner. In contrast to EET-induced systemic vasodilation [17,29], EETs elicit pulmonary vasoconstriction, responses that act as an initial step in evoking PAH and are significantly promoted by estrogen [10, 12]. As we demonstrated, high levels of pulmonary EETs caused by either estrogeninduced downregulation, or pharmacologic inhibition of sEH were proportional to the early occurrence and great increment of elevation of PA pressure (Figure 2,3), while decreases in pulmonary EETs via overexpression of sEH significantly attenuated RVSP (Figures 1-2). Moreover, the hypoxia pulmonary vasoconstriction (HPV) [30], represents an EET-potentiated pattern, as evidenced by significantly augmented HPV in mice with a knockout of the Ephx2 gene (sEH-KO) compared to their wild type (WT) controls. This enhanced HPV in sEH-KO mice was reversed by an EET antagonist, while restored in WT vessels treated with EETs [13,14,30]. It is worthy to note that even though the female-specific methylation of the Ephx2 gene, associated with higher circulating and tissue levels of EETs have existed in normal conditions, the basal PA pressure between sexes remains comparable (Figure 2a). This physiologically enhanced, yet dampened, action of EETs as a “first hit” becomes discernible only in the co-existence with pathological stimulus, such as SuHx, as a “second hit” that in most instances, seems to be a prerequisite for the development of clinical phenotype of PAH. Additionally, the EET-mediated elevation of PA pressure in the early stage of PAH was primarily elicited by decreases in EET degradation attributed to the female/estrogen-dependent methylation of the Ephx2 gene (Figure 6), and not caused by increases in EET synthesis because the CYP2C23 expression was comparable between sexes at that moment (Figure 3b). Ultimately, sex-differences in the incremental elevation of PA pressure disappeared, as the result of comparable increases in pulmonary EETs tween both sexes (Figures 4 & 5c). Thus, the next step was to identify a specific pathway(s) that directs the methylation-independent modulation of the Ephx2 gene, as a function of prolonged SuHx/or hypoxic pathology.
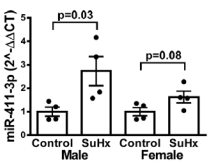
Figure 8: miR-411-3p levels in lungs of male and female rats before (normoxia) and after exposure to SuHx for 21 days, showing a SuHxinduced significantly increase in miR-411-3p mRNA expression in male rats. (n=4-5 with two-way ANOVA).
EET-dependent exacerbation of PAH development in males via the chronic hypoxia-stimulation of CYP2C23 expression and hypoxamir-dependent modulation of the Ephx2 gene
Chronic hypoxia-induced changes in sEH and CYP2C expression appear to be more evident in the pulmonary compared to the systemic circulation [14]. As reported, in multiple PAH models and patients with smoke-induced chronic obstructive pulmonary disease (COPD), pulmonary sEH expression was significantly attenuated, or undetectable in certain cases [31-33], whereas corresponding mechanisms remain unknown.We found that the methylation of Ephx2 gene was unaffected by hypoxia (Figure 7), the question thereby arose as to whether hypoxia can activate upstream-located regulatory miRNAs that in turn, target the Ephx2 gene to suppress its expression? As known, miRNAs are short non-coding RNAs that regulate gene expression posttranscriptionally. They generally bind to the 3’-UTR (untranslated region), of their target mRNAs and suppress protein synthesis by destabilizing the mRNA and translational silencing [34]. Indeed, hypoxia has been deemed as a key player in altering miRNA biogenesis and activity; the altered miRNAs, in terms of hypoxamirs, serve as repressors to modulate their target gene(s) expression under oxygen deprivation [23]. Figure 8 indicates that when PAH was established, the male-favorable increase in miR-411-3p serving as a hypoxamir, significantly suppressed the Ephx2/sEH expression, which eliminates sex differences in sEH expressions existed in the normal and early stage of PAH (Figure 3a). Of note, even though female SuHx rats exhibited statistically insignificant trends of increase in miR-411-3p expression (Figure 8), their sEH expressions were further decreased compared to normoxic controls (Figure 5a). In this regard, we interpret our data to mean that the hypoxamir-mediated responses cooperate with major actions of methylation of the Ephx2 gene, making the ultimate downregulation of sEH in female SuHx rats. Alternatively, in male SuHx rats, the hypoxamir-directed downregulation of Ephx2/sEH (Figure 5a), in combination of upregulation of CYP2C23 (Figure 5b), contributes primarily to increases in EETs (Figure 5c). To the best of our knowledge, the present study provides the first evidence indicating miR411-3p as a specific hypoxamir that uniquely targets the Ephx2 gene to suppress sEH expression during the progress of PAH. It is important to note that miRNAs target to gene(s) in a cell- or context-specific manner, and each specific gene can be targeted/ regulated by multiple miRNAs. In this context, the identification of miR-411-3p as a specific trigger for modifying the Ephx2 gene does not necessarily mean to exclude contributions from other hypoxamirs, but rather highlights the clinical significance for therapeutic inhibition of the hypoxamir(s) during PAH treatment, since we did find an upregulation of pulmonary miR-411-3p, concomitant with a downregulation of sEH expression in the lung of male PAH donators (date not shown). On the other hand, chronic hypoxia-stimulation of EET synthases [14,35,36], acts also in a male-predominant manner (Figure 5b), a phenomenon that we have no specific explanation for at the moment, but speculate to be correlated to estrogen-modulated hypoxic responses. For instance, hypoxia-inducible factors (HIFs) play key roles in the control of CYP gene expression under an oxygen deprivation condition. Certain transcription factors, such as NF-κB, are able to activate HIF-1β and HIF-α [37], whereas the IKK/ NF-κB signaling can be inhibited by estrogens. This may provide an alternative interpretation for the absence of upregulated CYP2C23 in female SuHx-PH rats.
SUMMARY AND PERSPECTIVES
The study has indicated two divergent regulatory mechanisms for epigenetic modulation of the pulmonary Ephx2 gene, via a female-favorable methylation-dependent pathway and a malepredominant hypoxamir-driven signaling to potentiate EETmediated elevation of PA pressure during different (early and later) stages of PAH. Although to date, there is lacking consensus for whether the increase in EET synthesis and decrease in EET hydrolysis are universally applicable to all types of PAH, our studies have demonstrated the intensified EET bioavailability in rats exposed to either combined (Sugen-hypoxia) or single hypoxia [14], a pathological pathway that emerges in most, if not all, types of PAH. Additionally, we have indicated miRNA-411-3p as an upstream modulator of the Ephx2 gene. Moving forward, further studies will provide functional evidence for the inhibition of miRNA-411-3p-dependent downregulation of sEH to prevent or alleviate EET-promotion of PAH, by in vivo administration of miRNA-411-3p specific siRNA to rats, followed by monitoring their PA pressure during SuHx exposure.
FUNDING
This work was supported by grants NIH HL070653, HL129797, HL144528, HL151187 and the biomedical and sciences research council of the Touro college and university system #171340.










































































































































{kind=link}