Epigenetic Mechanisms in Chikungunya Virus Infection and Their Role in Viral Pathogenesis
- 1. Department of Genetics, School of Medicine, Autonomous University of Nuevo León, Mexico
- 2. Department of Medical and Veterinary Entomology, School of Biological Sciences, Autonomous University of Nuevo León, Mexico
Abstract
The Chikungunya virus (CHIKV) is an alphavirus transmitted by mosquitoes. Patients infected with CHIKV usually develop an acute febrile illness after an incubation period of 5-7 days. The clinical features of chikungunya fever include high fever, maculopapular rash, headache, polyarthritis/arthralgias, myalgias, nausea, vomiting and diarrhea. Joint pain is limited to the peripheral joints and lasts for 7-10 days. Most infected people recover within weeks, but it has been reported that approximately ~25% of patients have had the disease for more than 2 months, about ~14% for more than 18 months, and up to ~5% remain symptomatic for 3-13 years after the initial infection, referring to these symptoms as chronic chikungunya arthritis (CCA). The present review addresses the epigenetics of CHIKV and its role in the pathogenesis of the virus, given the emergence and rapid dissemination of this condition that has become a public health problem worldwide, it is relevant to address to summarize the data known so far about CHIKV.
Keywords: Chikungunya; Alphavirus; Polyarthritis; Chronic Chikungunya Arthritis; Epigenetics
Introduction
The Chikungunya virus (CHIKV) is an alphavirus transmitted by mosquitoes. It has a single-stranded, positive-sense RNA genome of approximately 12 kb. It was first isolated from the serum of a febrile individual in Makonde, a province in southern Tanzania, in 1952, during a large outbreak of disease characterized by joint pain and high fever. Due to the stooped posture and stiff gait of infected individuals, the disease was named chikungunya, a word from the Bantu language of the Makonde ethnic group meaning “that which bends up” [1]. Since its description in 1952, CHIKV has caused millions of human infections in Africa, the Indian Ocean islands, Asia, Europe, and the Americas [2]. Like other arboviruses, CHIKV does not appear to affect the physical state of its arthropod vectors and is generally non-pathogenic, persisting for the vector's lifetime. In contrast, in vertebrate hosts, infections are typically pathogenic, with variable magnitude and duration depending on the peak viral concentration in the organism [3]. Other factors, such as viral adaptation, environmental and ecological conditions, and anthropogenic behavior, have favored its successful spread and persistence, negatively impacting public health and causing economic damage in affected regions [4]. In recent years, studies on resistance or susceptibility to infectious diseases have used classical biomarkers, such as single nucleotide polymorphisms (SNPs) [5]. However, emerging research highlights the importance of epigenetic alterations in immune cells as an equally relevant means of detecting and understanding the progression of infectious diseases [6,7].
Epigenetics in Host–Pathogen Interaction during Arbovirus Infection
The human genome comprises all human genetic information, while the epigenome describes genome modifications that determine whether genes are turned on or off depending on location and need, without altering the DNA sequence itself. Unlike genetic information, the epigenome is dynamic and responsive to external stimuli. Some of these stimuli can cause abnormal DNA modifications that, in turn, may alter normal gene expression and contribute to the development of various pathologies [8-10]. There are several types of epigenetic modifications, such as DNA and RNA methylation, histone modifications (acetylation, methylation), nucleosome positioning, and silencing associated with non-coding RNA [8-10]. These well-characterized epigenetic mechanisms influence gene availability for activation or suppression, which in turn affects gene expression patterns and key cellular processes in the host [11]. In the last decade, epigenetic research has rapidly advanced in understanding developmental biology, memory, and heritability [12]. More recently, it has become increasingly relevant in studies on oncology, as well as adaptive and innate immunity [13]. Although epigenetics has evolved in other fields, it has received comparatively little attention in the study of infectious diseases and host–pathogen interactions. Nevertheless, there is strong evidence supporting the involvement of epigenetics in host defense against pathogens in biological interactions, including DNA and RNA methylation, histone modification, and non-coding RNA activity [14]. Regarding epigenetic modifications caused by viral infections, reports suggest that DNA viruses play a significant role in host methylation by epigenetically deregulating cellular pathways to optimize their own transcription, replication, or evasion of the host’s innate immune response [15]. Parallel studies have provided growing evidence supporting the hypothesis that RNA viruses such as influenza, coronaviruses, and some flaviviruses are capable of reprogramming the host’s epigenetic landscape and controlling the innate immune system [8,16,17]. This leads to regulated changes in host gene expression that generate a favorable environment for completing their life cycles, evading host immune responses, and establishing a latent state, thereby creating conditions for persistent infection [13]. In this context, arboviral infections are among the leading public health problems worldwide. In particular, viruses from the Flaviviridae and Togaviridae families are generally pathogenic, often acute, and cause population-level viremia, representing a significant medical and economic burden for healthcare systems in endemic regions [4]. De Aguilar G et al., explains, “To date, some studies of epigenetic markers have investigated a series of epigenetic modifications, particularly miRNA/siRNA, iRNA suppression, modifications of iRNA regulators, and DNA methylation, in relation to host responses to arbovirus infections—mainly Dengue, Zika, Japanese encephalitis, Chikungunya, West Nile, and Crimean-Congo”[18].
Epigenetics in CHIKV
The nsP2 protein of CHIKV is a multifunctional protein involved in various processes of viral replication and evasion of the host immune response. This protein consists of five domains: an N-terminal domain, two RNA helicase domains of superfamily 1, a papain-like protease, and a domain similar to the FtsJ-like methyltransferase (MTase) [19,20]. The MTase domain has been considered non-functional as a methyltransferase due to the absence of key elements essential for this activity [19]. However, recent studies have shown that this domain is involved in the inhibition of interferon (IFN)-induced gene expression [20], suggesting a regulatory role in modulating the antiviral response. Although RNA methylation activity by the MTase of nsP2 has not been evidenced, this is a common function of methyltransferases that affects RNA stability and processing [21]. Another nonstructural protein, CHIKV nsP1, is an enzyme involved in protecting viral mRNA, thereby enhancing its stability. This enzyme comprises 535 residues that perform multiple functions, including 5' capping, interaction of the replication complex with the host cytosolic membrane, downstream translation regulation, and subgenomic RNA synthesis. It has two functional domains with methyltransferase (MTase, residues 28–259) and guanylyltransferase (GTase) activity [19]. These enzymes are largely responsible for installing and removing modifications in viral RNA, suggesting that post-transcriptional modifications may play essential roles in virus-host interactions [22]. Currently, over 170 post-transcriptional RNA modifications are known, now understood as a new level of regulation in gene expression control [23]. In RNA viruses, these modifications and their functions have been recognized for decades, but their biological significance in the host remains limited [22]. Several viruses with cytoplasmic replication cycles virally encode a methyltransferase protein that adds a guanosine cap, although the method varies among groups. Interestingly, alphaviruses, flaviviruses, and human immunodeficiency virus type 1 (HIV-1) contain not only the canonical m?G cap but also di- and tri-methylated guanosine caps [24].
A study by McIntyre et al., in ZIKV-, DENV-, HCV-, HPV-, and HIV-1-infected cells, showed that all five RNA viruses significantly altered the global post-transcriptional RNA landscape [24]. The most prevalent post-transcriptional RNA modification is N6-methyladenosine (m?A) in eukaryotic RNA. This modification has been associated in several viruses, including those mentioned above, as a potential regulator providing widespread control over viral life cycles and host-pathogen interactions [8]. Although there is no evidence of post-transcriptional modifications in CHIKV or any other alphavirus, this background suggests that a similar modification mechanism could exist. Similarly, epigenetic chromatin modifications-both in DNA and histones (methylation, acetylation, phosphorylation, and ubiquitination)—are involved in regulating genome structure and function. Evidence shows that some viruses (KSHV, EBV, HBV, HPV, HIV-1, HCMV) can alter the host cell epigenetic state by modifying histones or inducing expression of DNMTs and/or polycomb group (PcG) proteins [14,25]. However, data on histone modifications and DNA methylation are scarce and limited to a few cases. For example, a study using DENV showed that the capsid protein translocates to the nucleus and acts as a histone mimic, binding to core histones (H2A, H2B, H3, and H4) to form heterodimers and interfere with nucleosome formation, potentially affecting cellular homeostasis and promoting viral replication (Figure 1) [26]. A similar study found a histone H4-like motif in the capsid protein of yellow fever virus (YFV), mimicking H4 acetylation and interacting with the BRD4 bromodomain, which regulates gene expression and influences YFV replication [27]. Another study on DNA methylation showed that ZIKV alters the DNA methylome of neural progenitors, astrocytes, and differentiated neurons in genes related to brain disorders such as intellectual disability, autism, and schizophrenia, suggesting that ZIKV infection during fetal development may lead to late-onset neuropsychiatric complications [28]. These findings may indicate shared properties among arboviruses for epigenetically manipulating the host genome. In addition to chemical modifications of DNA and histones, other epigenetic mechanisms associated with viral infections involve non-coding RNAs (ncRNAs), which contribute to post-transcriptional gene regulation [29]. In recent years, regulatory RNAs such as long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) encoded by both host and pathogens (viruses, bacteria, fungi, parasites) have been shown to act as potent modulators of pattern recognition receptor (PRR) signalling, which is crucial for detecting microbial components and initiating innate immunity and inflammation, impacting pathogen life cycles and expression of inflammatory genes (Figure 2) [13]. In the case of arboviruses, some RNA viruses have evolved ways to exploit RNA degradation products and use them as important virus-derived ncRNAs with diverse biological regulatory functions [30]. miRNAs are the most studied epigenetic modification in CHIKV. A thorough review found six studies: one used primary cultures [31], three used human cell lines [32,34], and two used mouse cell lines [35,36].Two studies used high-density microarrays [31,32], two used low-density arrays (~760 miRNAs) [31,36], and the remaining two analyzed specific miRNAs: miR-124 [34], and miR-146a [33]. Studies using high and low-density microarrays observed altered expression of various miRNAs during early CHIKV infection, involved in apoptosis and JAK-STAT, MAPK, WNT, and TGF-β signaling pathways. The JAK/STAT pathway is a key signaling route in the IFN response to viral infection [13]. Additionally, CHIKV nsP2 is a potent inhibitor of IFN-induced JAK-STAT signaling [37]. In flaviviruses, miR-34 represses Wnt signaling and regulates the IFN response [38]. The TGF-β signaling pathway controls vital processes such as proliferation, differentiation, apoptosis, and has also been linked to modulation of various viral infections [39]. A study using TGF-β inhibitors showed increased CHIKV-mediated cell death compared to untreated cells, indicating that TGF-β production regulates CHIKV infection [32]. MAPK is another pathway stimulated by DNA or RNA viruses and is believed to play a role in their replication cycle [40]. Several miRNAs targeting MAPK pathway genes (MAPK1, MAPK8IP1, MAP3K14, MAPK9, MAP4K4, etc.) were downregulated during CHIKV infection, suggesting that CHIKV alters the host immune response by modulating miRNA expression that regulates key antiviral pathways [36].
In studies focusing on individual miRNAs such as miR-124-3p and miR-146a, findings showed modulation of viral production and inflammatory responses, respectively. Notably, miRNAs can have multiple targets. For example, miR-124-3p has 1,820 predicted direct targets according to Target Scan [41]. One of the involved pathways is fatty acid degradation and metabolism [42], suggesting that fatty acid synthesis is required for CHIKV replication [43]. Likewise, inhibition of miR-124 expression reduces CHIKV viral production in human cells [34].
Moreover, hsa-miR-124 is predominantly found in neurons and acts as a key negative regulator of neuroinflammation, downregulating TNF-α and MHC-II [44], which suggests a possible association with encephalitis during CHIKV infection [45]. On the other hand, miR-146a is a negative regulator of inflammation expressed mainly in neurons, microglia, and astrocytes, and is also induced by TLR signaling via NF-κB [44]. A study by Selvamani et al. investigated the role of miR-146a in CHIKV pathogenesis in human synovial cells and underlying inflammatory manifestations. miR-146a expression increased after CHIKV infection, resulting in downregulation of TRAF6, IRAK1, and IRAK2 and greater CHIKV replication in human synovial fibroblasts [33]. However, miR-146a is also present in other viral infections such as DENV, CCHFV, and JEV [18]. It has also been linked to attenuation of inflammation in diabetic wounds [46], as a critical immunomodulator in lupus [47], and in many other immune and non-immune inflammatory diseases [48]. Thus, it is more associated with its known anti-inflammatory role and its modulation of immune responses [49]. Therefore, miR-146a modulates signaling to reduce inflammatory responses in the central nervous system after NF-κB stimulation, helping to limit excessive neuroinflammation.
Figure 1
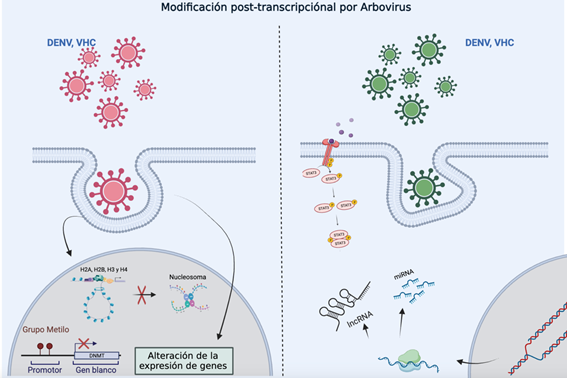
Figure 1: Representation of gene expression regulated by epigenetic modifications through miRNA or lncRNA. The diagram illustrates how the internalization of various viruses can induce post-transcriptional modifications by altering epigenetic processes such as promoter methylation, nucleosome modifications, or changes in miRNA and lncRNA expression, all of which directly impact the regulation of gene expression.
Figure 2
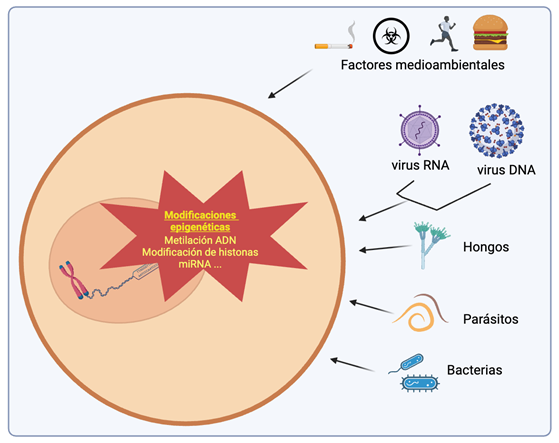
Figure 2: Schematic representation of the influence of various pathogens on epigenetic mechanisms. A wide range of pathogens—including viruses, bacteria, parasites, and fungi—can alter the host’s epigenetic mechanisms to promote their replication, persistence, or evasion of the immune system. These epigenetic modifications include DNA methylation, histone modifications (such as acetylation, methylation, and phosphorylation), and changes in the expression of non-coding RNAs such as miRNAs and lncRNAs. Such alterations can suppress genes involved in immune responses or activate genes that facilitate infection, thereby regulating gene expression without altering the underlying DNA sequence.
Final Considerations
Over the past decade, numerous studies have explored the epigenetics of virus–host interactions. These investigations have revealed that many viruses modulate their life cycle through epigenetic mechanisms, such as DNA or RNA methylation and chromatin modification. In addition, some viruses interfere with functions that antagonize the host cell’s epigenetic machinery, altering gene expression patterns and creating a favorable environment for viral replication and spread.
Studying epigenetics in the interaction between arboviruses and their hosts requires a range of complementary approaches to define and map the epigenetic landscape during infection. Thanks to technological advances, whole-genome analyses can now be performed using multi-omics techniques (epigenomics, transcriptomics, proteomics, etc.), enabling the integration of data and the identification of regulatory regions and genomic elements that play a crucial role in the epigenetic control of viral cycles. However, on a global scale, most research only superficially investigates the events occurring within infected cells. Therefore, continuous efforts are needed to gain a deeper and more comprehensive understanding of viral epigenetic control mechanisms in order to effectively combat complications associated with arboviral infections.
In this review, we have highlighted the scarcity of studies on the epigenetics of chikungunya virus infection and other arboviruses. It is clear that we are still in the early stages of understanding the substantial contributions of epigenetics to human disease and the shared response mechanisms across different arbovirus species. Much remains to be discovered about their overall impact. Ultimately, more research is needed to address virus–host interactions, leading to a clearer understanding of pathogenesis and disease development, as well as to the identification of potential therapeutic targets that may yield effective antiviral candidates—either alone or in combination with other therapies—in the future.
References
8. Gokhale NS, Horner SM. RNA modifications go viral. PLoS Pathog. 2017; 13: e1006188.
15. Milavetz BI, Balakrishnan L. Viral epigenetics. Methods Mol Biol. 2015; 1238: 569-596.
47. Shen N, Liang D, Tang Y, Qin Y. Chapter 5 - Epigenetics of Lupus. In: Wallace DJ, Hahn BH, eds. Dubois' Lupus Erythematosus and Related Syndromes (Eighth Edition). Philadelphia: W.B. Saunders; 2013: 46-56.









































































































































{kind=link}