Immunopathogenesis of Psoriasis
- 1. Dermatologist and Master of Science from the Medical College of the University of São Paulo, Medical Contributor To Severe Psoriasis Clinic of the Complexo Hospitalar Padre Bento de Guarulhos, Brazil
- 2. Dermatologist, Medical contributor to Severe Psoriasis Clinic of the Hospital Ipiranga, Brazil
- 3. Dermatologist of the Instituto de Assistência Médica ao Servidor Estadual do Estado de São Paulo, Brazil
Abstract
The immunopathogenesis of psoriasis involves complex interactions between the innate and the adaptive immune system. The central role for the TNF-α/IL-23/IL-17 pathway in the immunopathogenesis of plaque psoriasis has led to the development of successful therapies targeting these cytokines. Therefore, the knowledge of the main cells and cytokines involved in the pathogenesis of psoriasis is essential for dermatologists to understand better the disease as well as the mechanism of action of the biological therapies that have been revolutionizing the treatment of psoriasis in the last two decades. In this review the main and current knowledge of the immunopathogenesis of psoriasis is summarized.
Keywords
Immunity, Psoriasis, Cytokines, Interleukin-23, Interleukin-17, Tumoral alpha necrosis factor
Citation
Sanchez APG, Nunes DCB, Swiczar BCC (2020) Immunopathogenesis of Psoriasis. J Dermatolog Clin Res 8(2): 1131.
ABBREVIATIONS
TNF-a: Tumoral Alpha Necrosis Factor; IL: Interleukin; KC: Keratinocyte; PAMP: Pathogen-Associated Molecular Pattern; DAMP: Damage-Associated Molecular Pattern; PRR: Pathogen Recognition Receptor; TLR: Toll Like Receptor; ILC: Innate Lymphoid Cells; AMP: Antimicrobial Peptide; LL37: LL37/ Cathelicidin Molecule DC: Dendritic Cell; pDC: Plasmacytoid Dendritic Cell; INF: Interferon; mDC: Myeloid Dendritic Cell; Mφ: Macrophages; Th: Helper T Lymphocyte (CD4+ T Cell); Tc: Cytotoxic T Lymphocyte (CD8+ T Cell); NK: Natural Killer Cell; NET: Neutrophil Extracellular Trap; CXCL: Chemokine Ligand; ADAMTSL5: Metalloprotease Domain Containing Thrombospondin Type 1 Motif-Like 5; T0: Naïve T Lymphocyte; Ag: Antigen; PLA2G4D: Phospholipase A2 Group IVD; NKT: Natural Killer T Cell; TGF: Transforming Growth Factor; GMCSF: Granulocyte Macrophage Colony-Stimulating Factor; γδT: gamma delta T cell; T Reg: Regulatory T Cell; TRM: T Resident Memory Cell
INTRODUCTION
Psoriasis is a common chronic immune-mediated skin disorder that affects approximately 1,3% of the Brazilian population [1], and an estimated 125 million people worldwide [2]. Plaque psoriasis is the most common variant [2], and this review is focused on its immunopathogenesis.
In genetically susceptible individuals, psoriasis can be initiated or exacerbated by several environmental and behavioral factors including skin trauma, infections (e.g., streptococcal and human immunodeficiency virus infection), smoking, certain medications (e.g., lithium, beta adrenergic receptor blocking agents, antimalarials and interferon) and possibly stress [2,3].
Therefore, environmental stimuli and loss of tolerance lead to alteration of the innate and adaptive immunity, proliferation of keratinocytes (KCs) and the appearance of skin lesions [4-9].
ACTIVATION OF THE INNATE IMMUNE SYSTEM
The hallmark of plaque psoriasis is sustained inflammation leading to uncontrolled KCs proliferation and dysfunctional differentiation [4,6-9]. The beginning of the immune response is not fully elucidated [4,5,10]. The presence of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) in the epidermis (due to environmental factors) can lead to the activation of pathogen recognition receptors (PRRs) in KCs [4,5,10-12]. There is enhanced expression of toll like receptors (TLRs) in the KCs (TLRs are PRRs) of psoriatic patients [4,10-12]. The inflammatory process probably begins with the activation of TLRs (by PAMPs and DAMPs) in KCs, which produce several pro-inflammatory cytokines (TNF-α, IL1β) as well as antimicrobial peptides (AMPs) [4,5,11-13].
These AMPs (mainly LL37/cathelicidin molecules) are overexpressed in psoriatic plaques and protect the injured skin from infections [4-7,9-13]. However, LL-37 can bind to selfnucleic acids and trigger the activation of TLRs in DCs [4-7,9- 13]. Therefore, AMPs are probably involved in the initiation of inflammation in psoriasis by breaking innate tolerance to otherwise inert extracellular self-nucleic acids [4-7,9-13].
The release of LL37 by activated KCs and the subsequent binding of LL-37 to self-DNA in susceptible individuals might cause conformational changes to the bound DNA molecule, transforming this otherwise innocuous molecule into an aggregated, dense structure. LL-37 complexed with self-DNA is recognized by TLR9 in plasmacytoid dendritic cells (pDCs) [7,10-12], which accumulate in the dermis of early developing psoriatic lesions [10,13], and produce large amounts of IFN-α [2,4- 7,9,10,13]. IFN-α plays a critical role in the early phase of psoriasis pathogenesis as it triggers activation of myeloid dendritic cells (mDC) [10,13]. The number of mDCs is increased in psoriatic plaques and mDCs can be activated by many triggering factors, such as the skin microbiome, drugs, trauma, LL-37 complexed with self-RNA as well as by cytokines such as INF-α and TNF-α [4,10-12].
TNF-α is produced by several activated cells at the beginning of the inflammatory cascade and induces the maturation and activation of DCs [4,9,10,13]. In the presence of TNF-α, pDCs lose their ability to produce IFN-α and produce TNF-α [13]. Therefore, in classical plaque psoriasis, early transient overexpression of IFN-α is replaced by a dominant TNF-α driven chronic inflammation [13]. TNF-α is also produced by activated mDCs, macrophages (Mφ) and KCs [4,5,9].
It is important to note that the overproduction of TNF-α by several activated innate immune cells (pCDs, Mφ, KCs) drives the production of IL-23 (and other inflammatory cytokines) by mDCs [4-7,9,13]. Besides, TNF-α accelerates the infiltration of lymphocytes, monocytes and neutrophils from the peripheral blood into skin with DCs and KCs activation [4-7,9].
IFN-γ can also be found in psoriatic lesions and can promote IL-23 (and IL-1β) production by DCs and Mφs [5,7,9]. The main sources of IFN-γ are T lymphocytes and Natural Killer cells (NKs) and the production of INF-γ by these cells is driven by IL-12 [5,7,9].
Activated mDCs produce several pro-inflammatory cytokines (IL-23, TNF-α, IL-12, IL-6, IL-20) [4-7,9-12,14]. Among these cytokines, IL-23 is considered the key cytokine in maintaining the chronic inflammatory process in psoriasis plaques, as it stimulates many cells (of the innate and adaptive immune system) to produce IL-17 (or IL-17A) and other cytokines that maintain the exaggerated activation of KCs (Figure 1) [4-10].
In plaque psoriasis IL-23 is mainly produced by mDCs, but Mφ and KCs are also sources of IL-23. [4-10,15]. IL-23 activates innate immune cells (mainly ILC type 3 and γδ T cells) to secrete IL-17A and IL-22. In turn, IL-17A and IL-22 stimulate the production of chemokines, cytokines and AMPs by KCs, in addition to modify their differentiation and proliferation [4-7,9-12,14]. Therefore, at the beginning of the inflammatory cascade innate immune cells produce IL-17A and IL-22, even before the arrival and participation of conventional T lymphocytes (Th and Tc cells) in the immune response. The chemotactic proteins produced by activated KCs attract more DCs, monocytes, T lymphocytes, NKs and neutrophils which maintain the inflammation [4-7,9,10,14].
IL-17A is the major cytokine involved in the recruitment, activation, and survival of neutrophils in psoriatic lesions [4- 7,9,10,16]. The abundant presence of neutrophils in the skin lesions serves as a typical histopathologic hallmark of psoriasis (spongiform pustules of Kogoj and Munro’s micro abscesses) [14]. Neutrophils are sources of AMPs (LL-37), IL-17A and several other inflammatory mediators (reactive oxygen species, elastase, myeloperoxidase, proteinase 3, lipocalin 2, cathepsin G) [14,17]. The chromatin (DNA) of neutrophil extracellular traps (NETs) combined with LL-37 can contribute to activation of pDCs [14]. Proteinase 3 converts transmembrane TNF-α in KCs to soluble TNF-α (14) and proteinase 3, elastase and cathepsin G cleave proIL-36 (secreted by KCs) to activated IL-36 forms [7,9,14,17].
By the way, the members of the IL-36 family are highly expressed in plaque psoriasis [7,9,14,15,17] and their release from KCs is stimulated by IL-1β, TNF-α, IL-17A and IL-22 [7,9,14,15,17]. KCs secrete IL-36 cytokines in full-length forms (pro-IL-36 cytokines) which have minimal biologic activity [14,17]. When exposed to NETs and neutrophil proteases (or KCs derived proteases such as cathepsin S), pro-IL-36 cytokines are converted into shorter, active forms [14,15,17]. IL-36 cytokines amplify their own production in KC and induce expression of several pro-inflammatory molecules in KCs such as neutrophilic chemo-attractants (CXCL1, CXCL2, and CXCL8), AMPs (S100A7 and LL37) and IL-23 [7,9,14,15,17].
ACTIVATION OF THE ADAPTATIVE IMMUNE SYSTEM
Some activated mDCs end up capturing an autoantigen on the skin not yet defined (e.g., LL37; keratin 17; ADAMTSL5) [4- 6], and migrate to the regional lymph node, in search of naïve T lymphocytes (T0) capable of recognizing such antigen (Ag) [4,18]. Most recently, phospholipase A2 group IVD (PLA2G4D) was also reported as a possible psoriasis autoantigen. Its expression is increased in psoriatic KCs and mast cells and results in the generation of nonpeptide neolipid Ags presented on CD1aexpressing DCs for recognition by autoreactive T cells [5,6].
Upon recognizing the Ag, naïve T cells (T0) differentiate into specific subtypes, depending on the cytokines that predominate in the lymph node at the time of activation [18]. As previously reported, the activated mDCs are large producers of many cytokines (TNF-α, IL-12, IL-23, IL-6) [4,5,6,9-12,14]. These cytokines (along with others) lead T0 of psoriasis patients to differentiate preferably in T17, T22 and T1 [4-6,8,18]. It is important to note that the differentiation for subtype 17 is dependent on IL-6, TGF-β, IL-1 and IL-21; differentiation for subtype 22 depends on TNF-α and IL-6, while differentiation for subtype 1 is dependent on IL-12 and INF-γ [18].
The activated T lymphocytes leave the lymph node and when they reach the skin, Tc concentrate in the epidermis and the Th in the dermis [4,5,11,12,18,19].
In the dermis, activated Th lymphocytes again recognize the Ag and, in the presence of IL-23 (Th17 and Th22) and of IL-12 (Th1), Th cells proliferate and produce their respective cytokine repertoires (Th17: IL-17A, IL-17F, IL-22, TNF-α, IL-26, IL-29, GMCSF; Th22: IL-22, TNF-α and Th1: INF-?, TNF-α) [4-9].
Years ago, the maintenance of the chronic inflammatory process in psoriasis plaques was associated with the excessive production of IL-12, formed by two protein chains (p40 + p35) [5-7,9]. DCs, Mφ, and neutrophils produce IL-12 and the binding of IL-12 to its receptor (on Th1 and NK), triggers the production of INF-? [9]. IFN-γ enhances Ag processing and presentation and drives Th0 cells towards a Th1 response [5,6,9]. So, the identification of high p40 concentrations in psoriatic lesions suggested that IL-12 would be a key cytokine in plaques, stimulating the proliferation of Th1 and their production of INF-γ (and TNF-α) [5-7,9]. However, the discovery of IL-23 (which also contains p40 in its molecular structure, being formed by p40 + p19) led to further studies, demonstrating that in psoriasis plaques there are high concentrations of p19 in association with p40 in the lesions, i.e. IL-23 (p19 + p40) [5-9].
As previously mentioned, in addition to dermal DCs, KCs and Mφ produce IL-23 [4,5,7-10,15], and nowadays IL-23 is considered the key cytokine in maintaining the chronic inflammatory process of psoriasis, as it stimulates many immune cells (e.g., Th17; ILC type 3 and γδT cells) to produce IL-17A (Figure 1) [4,7,8,9,20].
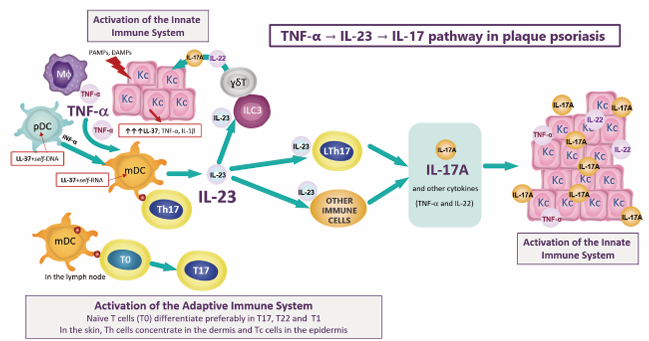
Figure 1 TNF-α/IL-23/IL-17 pathway in the immunopathogenesis of plaque psoriasis. PAMPs: pathogen-associated molecular patterns; DAMPs: damage-associated molecular patterns; LL-37: LL37/cathelicidin molecule; KC: keratinocyte; pDC: plasmacytoid dendritic cell; mDC: myeloid dendritic cell; Mφ: macrophage; γδT: gamma delta T cell; ILC3: innate lymphoid cell type 3; Th: helper T lymphocytes (CD4+ T cell); Tc: cytotoxic T lymphocytes (CD8+ T cell); T0: naïve T lymphocyte; IL: interleukin; TNF-α: tumoral necrosis factor alpha; INF-α: interferon alpha; OTHER IMMUNE CELLS: Tc17, ILC3, γδT, neutrophils, mast cells, natural killer cells, natural killer T cells.
IL-17A, alone and in additive/synergist combinations with cytokines such as TNF-α, IL-22, INF-? and IL-17F, is a key factor for altering responses of KCs. Therefore, IL-17A and other cytokines (produced by Th17, Th22 and Th1) induce the expression and release of many psoriasis-related proteins from KCs (cytokines, AMPs, chemokines and growth factors) and also interfere in the differentiation and proliferation of KCs (Figure 2) [4-10,17,19,21].
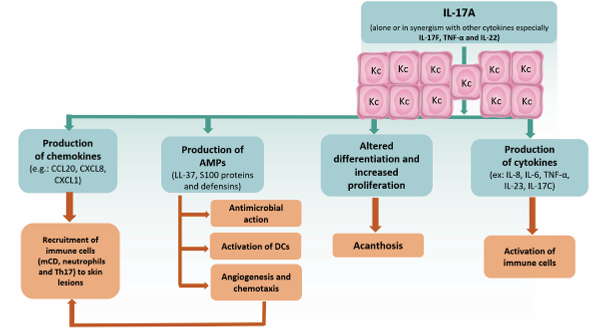
Figure 2 Potential roles of interleukin (IL)-17A in the pathogenesis of plaque psoriasis. IL-17A acts on keratinocytes (KCs) to stimulate production of AMPs, proinflammatory chemokines and cytokines. DCs: dendritic cells; AMPs: antimicrobial peptides.
As the IL-23/IL-17 axis maintains the chronic inflammatory process in plaque psoriasis, Th17 are the most important adaptive immune system cells in the immunopathogenesis of the disease [8,9]. In the presence of IL-23, Th17 increase their expression of IL-23 receptors (become more responsive to IL-23), and produce large amounts of IL-17A and others cytokines (IL-17F, IL-22, GMCSF, TNF-α, IL-26, IL-29) [5-7,9,10].
Basic research and several clinical trials have suggested a dominant pathogenic role in patients with psoriasis for IL17A [21]. IL-17A autoactivates its own synthesis in Th17 and in synergy with other cytokines (e.g., TNF-α, IL-1β and IL-17F) induces many inflammatory products in KCs, including IL-17C, pro-IL-36 cytokines, CXCL8/IL-8, IL-19 and AMPs [21]. Besides, IL-17A acts on non-immune cells and alters the biology of many cell types involved in the genesis of systemic inflammation and associated comorbidities [5,9,16,20,22,24].
IL-17A and IL-17F share the highest structural homology inside the IL-17 cytokine family and by this fulfill very similar biological functions [16,20,24]. IL-17A and IL-17F are mainly produced by immune cells and strongly contribute to tissue inflammation. [16,20,24]. IL-17F is less potent than IL-17A, despite being 50% homologous [16,20]. In psoriatic KCs, IL17F together with IL-17A induce overproduction of many inflammatory cytokines, chemokines and AMPs [16,20,21]. IL17A and IL-17F stimulate KCs to produce IL-17C which binds to its receptor expressed on Th17 cells and KCs [14,16,21,23]. IL-17C maintains an autocrine loop in KCs and stimulates Th17 cells to secrete IL-17A [14,21,23]. IL-17E, another member of the IL-17 family, contributes to the infiltration of neutrophils in the psoriatic lesions [14,16].
IL-26 and IL-29 produced by Th17 cells upregulate KCs expression of CXCL9, CXCL10, and CXCL11. These chemokines(and IL-12) promote the recruitment of Th1 lymphocytes, which can be found in well-developed psoriatic plaques [8].
IL-17A, IL-17-F and TNF-α induce the production of IL19 and IL-36 by KCs [8,6,20,21]. IL-36 acts cooperatively with IL-17A to promote epidermal proliferation and disrupt the normal differentiation of KCs [8,21]. IL-22 (produced by Th17 and Th22 cells), and IL-19 stimulate KCs hyperproliferation and the upregulation of proinflammatory signals, including AMPs [8]. Thus, IL-22, IL-19 and IL-36 contribute to epidermal hyperplasia and altered terminal differentiation, which lead to the characteristic thickening of thickening of the skin seen in patients with chronic psoriasis [6,8,20,21].
In addition to acting on Th17, IL-23 stimulates the release of IL-17A by others cells from both the adaptive immune system (Tc17, regulatory T cells – Treg), and innate immune system (ILCs type 3, γδ T cells, NKs, NKTs, neutrophils and mast cells) [4-7,9,16,20,23-26]. Therefore, many cells are sources of IL-17A in psoriatic lesions.
It is important to mention that the function of T reg is impaired in psoriasis [10,23,25]. Interestingly, in the presence of IL-23, T regs of patients with psoriasis have an enhanced propensity to differentiate into IL-17A-producing cells [23,25].
Another important issue is the participation of T resident memory cells (TRMs), in the inflammatory cascade. TRMs are enriched in active and resolved psoriatic lesions, thereby influencing the onset and maintenance of a psoriatic plaque [27]. Upon disease triggers, Langerhans cells and TRMs actively produce proinflammatory cytokines (IL-23, IL-17A and IL-22), and can cause recurrent inflammation [27].
In conclusion, psoriasis is a chronic inflammatory skin disease that involves numerous types of immune cells and cytokines resulting in an inflammatory feedback loop and hyperproliferation of the epidermis (Figure 1 and 2).
DISCUSSION AND CONCLUSION
The main cytokines involved in the immunopathogenesis of psoriasis are TNF-α, IL-23 and IL-17A (Figure 1). TNF-α participates in all stages of the immunological cascade of psoriasis, but its main action is to maintain the activation of mDCs. Excessive production of IL-23 by mDCs greatly amplifies the inflammatory cascade, since IL-23 stimulates the production and release of IL-17A by Th17 and Tc17, as well as by several cells of the innate immune system (Figure 1) [4-9,16,20,23-27]. IL-17A acts alone or synergistically with other cytokines (e.g., TNF-α, IL-17F, IL-22) to induce the expression and release of many psoriasis-related proteins from KCs (Figure 2).
TNF-α, IL-23 and IL-17A excessively produced in psoriasis lesions, can reach the bloodstream and produce metabolic changes and vascular inflammation [2,20,22]. In other words, the same cytokines that drive the immunopathogenesis of psoriasis are involved in the immunopathogenesis of atherosclerosis and various comorbidities associated with psoriasis (obesity, liver steatosis, psoriatic arthritis and inflammatory bowel disease) [2,20,22]. Therefore, plaque psoriasis should be considered, especially in its most severe forms, a systemic disease [2,16,20,22].
The biological drugs that inhibit TNF-α, IL-23 and IL-17A have been used successfully and have revolutionized the treatment of plaque psoriasis in recent years, which corroborates the importance of these cytokines in maintaining the inflammatory process [5-9]. Finally, it is important to mention that studies with IL-23, IL-17A and TNF-α inhibitors are being carried out to deliver important insights into the role that biological agents may play in treating the systemic inflammation associated with psoriasis [8,20,22].









































































































































