Modeling of the Antiviral Peptide for Specific Inhibition of Influenza Virus Entry
- 1. Department of Chemistry and Biochemistry, Mendel University in Brno, Czech Republic
- 2. Central European Institute of Technology, Brno University of Technology, Czech Republic
Abstract
Influenza is a highly pathogenic virus well known by a higher mutation rate, resulted from antigenic drift and shift of its surface proteins. The hemagglutinin- influenza major virus surface protein is responsible for viral entry through targeting sialic acid present on the lipid membrane of the host cell. Nowadays, peptides become the perspective therapeutic agents for various infection diseases including influenza. Utilization of peptides could provide high specifity, attained by a number of possible modifications, which can be simply proposed by using in silico modeling.
Molecular modeling of peptides with high affinity towards sialic acid binding site of influenza hemagglutinin may be thus helpful for inhibition of viral entry. Hence, we employed Yasara program for molecular modeling to estimate a binding energy of selected peptides to specific hemagglutinin binding site in the amino acids range of 116 – 261. We designed a mimicking peptide with sequence WLVFFVIFYIFR showing binding energy 412.669 kcal/mol, which could be suitable to inhibit the influenza infection.
Keywords
• Hemagglutinin
• Influenza virus
• Peptide modeling
• Viral entry inhibition
Citation
Bastl K, Skalickova S, Heger Z, Zitka O, Adam V, et al. (2015) Modeling of the Antiviral Peptide for Specific Inhibition of Influenza Virus Entry. J Drug Des Res 2(1): 1010.
ABBREVIATIONS
HA: Hemagglutinin; NA: Neuraminidase
INTRODUCTION
Influenza is a highly pathogenic, respiratory disease caused by RNA influenza virus belonging to Orthomyxoviridae family. As the other RNA genome viruses, influenza virus undergoes high mutation rates and frequent genetic reassortment leading to variability in viral surface antigens hemagglutinin (HA) and neuraminidase (NA). While hemagglutinin helps to host cell entry, the neuraminidase is responsible for virus release from a host cell [1]. Both of antigens show potential to be an important target for antiviral drugs. Antiviral peptides seem to be helpful due to their effective antiviral properties, relative cost-effective production, rapid elimination following treatment and low levels of possible side effects [2]. The possibilities of therapeutically targeting brings on vary of their structure e.g. posttranslational modifications, point-mutations or conformational changes and possibility to utilize nano carriers to protect a peptide degradation in bodily internal environment [3]. Hence, the rational drug design using molecular modelling methods has become a highly efficient tool for development of the new therapeutic agents [4]. Also, the potential toxicity of novel antiviral peptides could be explored by preliminary studies [5]. The aim of our work was to suggest an antiviral peptide with potential activity to inhibit influenza virus entry into a host cell. The peptide sequence was designed with high affinity to sialic acid binding site of influenza hemagglutinin. It is known, that sialic acid, naturally present on host cell membranes, is responsible for hemagglutinin attachment to cell followed by viral fusion. Although, the same principle is applied by several antivirals drugs, the peptides provide reduction of viral resistance towards treatment. We think that by blocking the channel inside the HA1, by which it is bound to sialic acid receptor on the host cell membrane, we can block the binding or, at least reduce the affinity of HA, and thus inhibit the virus fusion to the host cell.The size of the peptide was chosen somewhat randomly, but the criteria were to best fit into the size of the active size of the HA1, which is between 10 to 15 A. We do not want to have too short peptide which might not to be specific enough, on the other hand, the peptide which would be too long, could create unfunctional tertiary structures before it reaches the target. Based on recent findings the peptides entry inhibitors should be rich in aromatic amino acids and strongly hydrophobic. Our assumption is that active site inside the HA creates almost straight hydrophobic channel, so the peptide must be conformationally flexible and hydrophobic with positively charged end, which directs the peptide out of the channel [6].
MATERIALS AND METHODS
All computations were performed in YASARA STRUCTURE (www.yasara.org) using the crystal structure of the hemagglutinin (PDB: 1RUZ). The sequence of P1WLVFFVIFYFFR andP2 WLVFFVIFYIFR were also generated to evaluate the robustness of the ligand binding to hemagglutinin.The docking results of each independent run were then treated with the docking analysis macro (Docking_EM_analysis S1) calculating the potential binding energy of the ligand, using default docking parameters and point charges assigned according to the AMBER03 force field. All dockings were performed after the short molecular dynamic simulation of either HA and the possible ligands in the water at pH 7.4 were run. The structures were cleared from water molecules and all necessary hydrogen was added. The simulations were run in periodic boundary conditions, with long range force cut off at 7.84 A. The docking was performed in the box of the length of 15 A around the active side of HA1, residues 116-261, which was confirmed by the docking of the sialic acid beforehand. The binding energies were calculated after splitting the ligand and the rest of the objects, against the rest of the soup. All atoms were surrounded by the box without periodical boundary conditions.
RESULTS AND DISCUSSION
Computational docking allows the fast screening of a large number of candidate ligands, which may afterwards be analyzed through more demanding computational techniques in the search for suitable leads for further development and experimental characterization. In our study, we selected peptide (P1)WLVFFVIFYFFR [7] with antiviral activity against influenza virus and change one amino acid in the chain WLVFFVIFYIFR (P2)to obtain higher hydrophobicity. Using molecular modeling we obtained, P1 and P2 bind preferentially to an exposed pocket in hemagglutinin subunit (Figure 1A) formed by the globular domain consists of HA1 residues 116–261 folded into a jelly-roll motif of eight stranded antiparallel β-sheets which is binding pocket for sialic acid (Figure 1B).
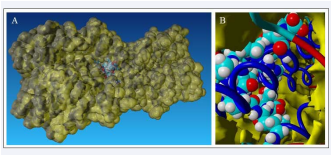
Figure 1: Peptide (P2) bound to sialic acid binding site of hemagglutinin: A) General view to exposed pocket in hemagglutinin subunit, B) Detailed view on peptide – hemagglutinin interaction.
Binding energies of each peptide to hemagglutinin were 372,279kcal/mol for P1 and 412,669 kcal/mol for P2. Thus, P2 peptide is potentially more suitable antiviral agent against influenza virus due to its hydrophobicity [8], and higher binding energy.
CONCLUSION
Our computational study confirms that P1 and P2 peptides are able to bind to the specific binding site of hemagglutinin and P2 shows higher binding energy and is more suitable for inhibiting influenza virus entry to the host cell by binding to the sialic acid binding pocket. This preliminary study will be used for further in vitro and in vivo experiments.









































































































































