A Case of Recurrent Cerebral Oedema in Diabetic Ketoacidosis
- 1. Department of Endocrinology, Children’s University Hospital, Temple Street, Dublin, Ireland
- 2. Department of Radiology, Children’s University Hospital, Temple Street, Dublin, Ireland
Abstract
A 7-year-old girl presented with new onset type 1 diabetes, with moderate diabetic ketoacidosis. She developed clinical and radiologically confirmed cerebral oedema managed with mannitol. Despite slow normalisation of biochemical parameters and marked clinical improvement, she had a further episode of symptomatic cerebral oedema 50 hours following presentation.
Keywords
• Diabetic ketoacidosis
• Cerebral edema
Citation
Hawkes CP, Laffan EE, Murphy NP (2013) A Case of Recurrent Cerebral Oedema in Diabetic Ketoacidosis. J Endocrinol Diabetes Obes 1(1): 1001.
ABBREVIATIONS
DKA: Diabetic Ketoacidosis; GCS: Glasgow Coma Scale; PICU: Pediatric Intensive Care Unit
INTRODUCTION
The incidence of diabetic ketoacidosis (DKA) at diagnosis of type one diabetes varies from 15 to 70%, and is more common at presentation in younger children, newly diagnosed (compared to established) diabetes, and those in lower socioeconomic classes [1]. Cerebral oedema is a significant cause of mortality in children with DKA and close observation for the development of this complication is required. We present the case of a 7 year old girl who developed a recurrence of symptomatic cerebral oedema despite apparent clinical recovery from her initial episode.
CASE REPORT
A 7 year-old previously well girl presented to the Emergency Department with a one week history of polydipsia and polyuria. She also had a 3 day history of vomiting and abdominal pain. On arrival, she was sleepy but rousable and had a Glasgow Coma Scale (GCS) score of 15/15. Her respiratory rate was 36, heart rate was 140, and capillary refill time was 3 seconds. Her venous blood results were consistent with moderate diabetic ketoacidosis (Glucose = 28.6mmol/L (515mg/dL), Ketones = 3.9mmol/L, pH = 7.12, pCO2 = 3.1kPa, HCO3 - = 9.2mmol/L, Base Excess = -20.3, Corrected Sodium = 130mmol/L, Potassium = 3.9mmol/L, Urea = 9.8mmol/L, Creatinine = 83umol/L). She was managed as per accepted guidelines [1]. She initially received 10ml/kg 0.9% NaCl over 30 minutes and was then maintained on 0.9%NaCl to correct an 8% deficit over 48 hours, with the inital bolus subtracted from fluid replacement. Insulin infusion was deferred for 60 minutes. Forty five minutes after starting intravenous fluids and prior to commencing insulin, she became acutely disorientated, confused and aggressive. This was followed by lethargy and poor responsiveness over a period of 5 minutes. Her GCS fell to 7. She was intubated and received 5ml/kg mannitol 20% for presumed cerebral oedema. The 8% fluid deficit was recalculated to replace over 72 hours. A non-contrast CT brain was performed following stabilisation. This showed effacement of the quadrigeminal plate, ambient and interpenduncular cisterns, as well as the cerebral sulci, with preservation of grey/white matter differentiation (Figure 1a and 1b).
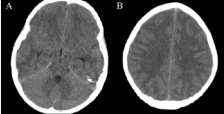
Figure 1 Five hours following presentation. Non contrast axial CT through the brain in e uncus and parahippocampal gyrus (A) There was also effacement of the cerebral sulci superiorly (B) All suggestive of diffuse cerebral edema.
A diagnosis of cerebral oedema was made.
Intravenous insulin was commenced at 0.1u/kg/hr after one hour of intravenous fluids. Intravenous fluids were initially 0.9% NaCl. Dextrose 5% was added to fluids at 5 hours to maintain normoglycaemia and 0.9% NaCl was changed to 0.45% NaCl at 18 hours. Insulin dose was titrated to 0.05 u/kg/hr at 15 hours. Extubation was attempted at 5 hours, but was postponed due to increasing agitation. A planned repeat dose of mannitol was given at 6 hours. Morphine and midazolam was continued until 20 hours, and was weaned prior to successful extubation. Her neurological examination was normal. Her pH, glucose, corrected sodium, osmolality and electrolytes remained within acceptable limits throughout management (Figure 2).
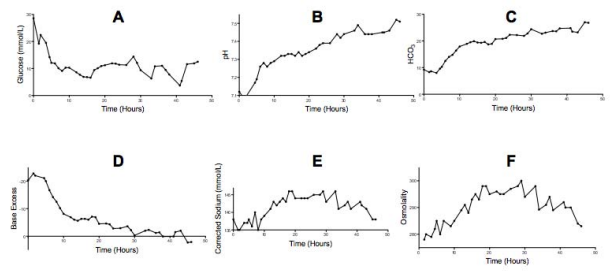
Figure 2 Slow normalisation of Glucose (A); pH(B); HCO3 (C); Base Excess (D); Corrected Sodium (E); and Osmolality (F).
At 45 hours, she had fully recovered clinically and was commenced on subcutaneous insulin before breakfast and intravenous insulin was discontinued at 46 hours.
At 50 hours, she became acutely less responsive. She initially localised to pain and speech consisted of inappropriate words (GCS 9). A futher dose of intravenous mannitol 20% (5ml/kg) was administered. Her GCS returned to 15 within half an hour. An MRI brain was subsequently performed (Figure 3).
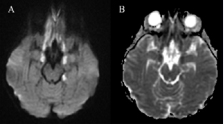
Figure 3 Axial diffusion weighted image through the base of the brain, demonstrates abnormal bright signal in the inferior frontal lobes and uncal regions bilaterally, and left hippocampal body (A) Theseabnormalities were also dark on the ADC map (B) Confirming restricted diffusion and acute irreversible cytotoxic edema.
This showed areas of abnormal T2 and T2 FLAIR, signal in several regions in the base of the brain, including the inferior frontal lobes posteriorly, the uncus and amygdala of the mesial temporal lobes bilaterally, both occipital lobes antermoedially and in the splenium of the corpus callosum. These areas showed restriction on diffusion-weighted imaging. The ventricles, cisterns and sulci looked patent and there was no cerebellar herniation. This distribution pattern was considered to be secondary to mass effect due to acute cerebral oedema, which had resolved by the time of the MRI. A repeat MRI brain was performed 2 weeks later showed improvement but not complete resolution of the above findings. She made a full recovery. There are no neurological sequellae at follow up three years after this event.
DISCUSSION
The annual risk of DKA in children with established type 1 diabetes is 8%, with risk factors including poor metabolic control, reduced access to medical services, pubertal females and psychiatric disorders [2]. At least two thirds of diabetes related mortality is related to DKA, with over two thirds of those being due to cerebral oedema [3]. Up to 40% of children who have had cerebral oedema have subsequent neurological sequelae [4,5]. Despite the increased morbidity and mortality associated with cerebral oedema in DKA, the aetiology is poorly understood. The majority of cases of cerebral oedema occur within 14 hours of treatment initiation [6], often after a period of clincial improvement. Some degree of increased intracranial pressure, manifested as ventricular narrowing, occurs in up to half of children with DKA [7]. but what causes a small proportion of those develop clinical cerebral oedema remains largely unknown. New diagnosis [4], increased serum urea nitrogen and reduced partial pressures of arterial carbon dioxide at presentation [6] convey an increased risk of developing cerebral oedema. Treatment with bicarbonate [6] or profound falls in serum osmolality [8] also increase the risk. Rapid rehydration [9,10] is also conisidered to increase the risk, and generally accepted management guidelines [1,11] mandate slow rehydration. Whether or not slower rehydration regimes have a protective effect is unclear and small studies suggest no difference in subclinical cerebral oedema between slow and more rapid rehydration [12]. The early onset of cerebral oedema following commencement of therapy may be a consequence of altered cerebral metabolism in DKA, followed by possible cerebral hypoperfusion and reperfusion injuries with treatment [13]. While the majority of cases occur soon after commencing therapy, recurrence following successful therapy has not previously been reported. In the case described, a 7 year-old girl with moderate DKA developed clinical and radiological cerebral oedema soon after commencing mangement in line with clincial guidelines. Despite careful appropriate normalisation of biochemical parameters, she had a second acute episode of deterioration at 50 hours.
The aetiology of this unexpected recurrence is unknown, and further highlights our poor understanding of the pathogenesis of this serious complication. Vasogenic cerebral oedema is associated with an elevated diffusion coefficient on diffusion weighted MRI, and this is seen during typical cerebral oedema in DKA. In this patient’s second episode, there was decreased diffusion on MRI. This suggests cytotoxic oedema and may have been a response to injury or ischaemia from the first episode.
While the trigger for her clinical deterioration is unclear, this case highlights the need for ongoing close observation following clinical improvement. Close observation should continue for 72 hours, and consideration given to reimaging if there are any concerns. In this case, the restricted diffusion on the initial MRI would suggest that the abnormalities secondary to the cerebral oedema were irreversible. This emphasizes the seriousness of this complication and the need for medical vigilance to detect and treat this condition. Fortunately there appear to be no long term sequelae at three years follow-up.









































































































































