Metformin in Cancer: Chemical Pathways for Tumoral Control Independent of Amp-Dependent Kinase
- 1. Endocrine and Metabolic Diseases Research Center. School of Medicine, University of Zulia, Venezuela
Abstract
Dimethylbiguanide (Metformin) is an anti-hyperglycemic drug used in the management of insulin resistance-related diseases like type 2 diabetes mellitus for over 40 years. The molecular mechanisms of action related to its metabolic effects are linked to dependent and independent AMP-dependent kinase (AMPK) activation. Epidemiological evidence has suggested that metformin administration has been associated to lower cancer risk and cancer-related mortality in patients with type 2 diabetes mellitus. Anti-tumoral properties have been described via AMPK-dependent and independent pathways. Metformin is a cationic molecule capable of copper sequestration and subsequent mitochondrial electron transport inhibition, compromising oxidative phosphorylation. Moreover, metformin has also been confirmed to modulate pluripotency in cancer stem cells, blunting their survival and proliferation. Finally, the reduction of vitamin B12 availability has also been suggested to control one-carbon metabolism, DNA synthesis and cytostatic effects. The purpose of this review is to examine the AMPK-independent related mechanisms concerning tumor expansion control and survival.
Keywords
• Metformin
• Biguanides
• Cancer
• AMPK
• Copper sequestration
• Mitochondrial toxicity
• Vitamin B12
Citation
Rojas J, Chávez-Castillo M, Torres W, Arraiz N, Cabrera M, et al. (2014) Metformin in Cancer: Chemical Pathways for Tumoral Control Independent of Amp-Dependent Kinase. J Endocrinol Diabetes Obes 2(2): 1036.
INTRODUCTION
Metformin is one of the most frequently prescribed drugs for insulin resistance-related conditions, such as Type 2 Diabetes Mellitus (T2DM) [1] and Polycystic Ovary Syndrome [2]. This medication has proven to enhance insulin sensitivity in liver [3] and skeletal muscle [4], improving metabolic control in these patients. The success of this outstanding drug relies on the activation of AMPK-dependent pathways [5] which result in greater glucose uptake [6], modulation of lactate production [7], inhibition of gluconeogenesis [8] and protein synthesis [9], activation of fatty acid β-oxidation [10], and central regulation of appetite [11]. Successful management of diabetic patients with metformin has been observed for over 20 years [12,13], being particularly preferred as first line of treatment as it allows inhibition of gluconeogenesis and improvement of impaired fasting glucose [14,15] while being less prone to inducing hypoglycemia, in comparison to other oral antidiabetic agents [16].
The relationship between diabetes and cancer is a two-way street. Johnson and Pollak have presented interesting insight explaining the intricate biological relationship between T2DM and inflammation-related neoplasia, as in colorectal, breast, and pancreatic cancer [17]. The mixture of a poor quality diet –low in antioxidants and rich in calories– associated with physical inactivity is related to hyperinsulinemia and insulin resistance. In turn, these factors have been related to development of obesity, inflammation and cancer [17,18]. Various meta-analyses have related diabetes with risk of several types of cancer after adjusting covariables, including bladder (RR=1.24; 95% CI: 1.08-1.42) [19], endometrial (RR=2.10, 95% CI: 1.75-2.53) [20], breast (RR=1.24; 95% CI:0.95-1.95) [21], hepatocellular (RR=2.5; 95% CI:1.9-3.2) [22] and even non-Hodgkin Lymphoma (RR=1.18; 95% CI:1.30- 2.47) [23], as well as higher mortality risk from colorectal cancer: RR=1.26, 95% CI: 1.05-1.50 [24]. In a population–based cohort study with over 65 thousand patients with no previous history of diabetes or cancer, Lin [25] reported that the greatest decrease in cancer risk was observed in those patients that had more than 1080 Defined Daily Doses, with a hazard ratio of 0.27 (95%CI 0.09-0.84), conferring a dosage-dependent effect to this drug. R
ecent evidence has suggested that T2DM patients who are long-term metformin users have lower risk for breast cancer. In a nested case-control study with T2DM patients over 50 years of age, Bosco [26] found long-term metformin users less likely to be diagnosed with breast cancer (OR=0.77; 95% IC: 0.61- 0.99). Likewise, Little [27] reported a borderline-significant negative association between metformin use and development of pancreatic cancer (OR=0.28; 95% CI: 0.06-1.22, p=0.09); which was absent in the group using sulphonylureas (OR=0.38; 95% CI: 0.08-1.70, p=0.20). These results were later supported by findings from Soranna [28]. Furthermore, a meta-analysis by Noto [29], ascertained a pooled risk ratio of 0.68 (95% CI: 0.53- 0.88) for all-cancer incidence and 0.66 (95% CI: 0.49-0.88) for cancer mortality, suggesting that metformin in T2DM patients lowered the risk for cancer development, including colorectal, liver and lung cancer.
Although current epidemiological evidence requires improved methodology in order to avoid time-related bias [30], this data have led to multiple in vitro and animal studies that appear to confirm the anti-tumoral activity of metformin, founded in various mechanisms dependent and independent of AMP kinase (AMPK) activity. The purpose of this review is to explore the anti-tumoral AMPK-independent properties of metformin, and correlate their efficacy against tumor cell metabolism.
PHARMACODYNAMICS AND PHARMACOKINETICS OF METFORMIN
Metformin, (N,N-Dimethylimidodicarbonimidic diamide or dimethylbiguanide), is a molecule belonging to biguanide family of drugs, very well-known for its hypoglycemic effect in animals since 1929, which was later confirmed in humans along with its sister drug, phenformin (phenylethyldiguanide) in 1957 [31]. In 1960, Kruger [32] pinpointed biguanides to exert their effects through modulation of carbohydrate metabolism and lowering oxygen uptake, inducing a pseudoanoxia state, which in turn induces anaerobic glycolysis, lactate production and lower glycemia. Additionally, these effects are observed solely in individuals with altered glucose metabolism –and not in healthy subjects– and they don’t appear to involve stimulation of insulin secretion [33].
Dimethylbiguanide is a hydrophilic molecule with positive charge at physiologic pH, which is not metabolized during its passage through the liver and circulatory system [34]. It has a molecular weight of 129,164 g/mol, with 50±16% bioavailability a half-life of ~5 hours and a renal clearance rate of 510±130 mL/min in subjects with appropriate kidney function [35]. Therapeutic levels of the drug range between 0.5-1.0 mg/L [34], with a threshold of 2.5 mg/L for lactic acidosis [35].
Absorption and distribution of metformin rely on a series of bidirectional transporters from 3 protein families in the intestine, liver and kidney [36,37]; Figure 1.
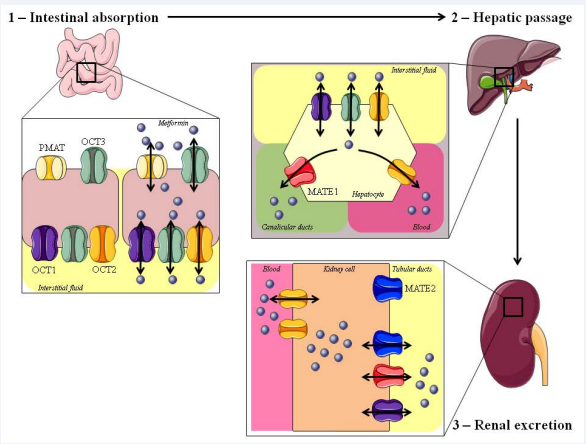
Figure 1: Absorption and distribution of Metformin. Metformin enter using the PMAT (Plasma Membrane Monoamine Transporter) and the OCT3 (Organic Cation Transporters) localized in the apical membrane of the enterocyte. They enter lymphatic circulation and the hepatocyte with the OCT1/2/3. Circulatory system is achieved via OCT2, as well as entrance to the kidney cell. Excretion towards tubular ducts is done with the MATE1 (Multidrug and Toxicity Extrusion protein), MATE2 and OCT1.
Translocation to the enterocyte depends on two transporters: PMAT (Plasma Membrane Monoamine Transporter, SLC29A4; OMIM 609149) and OCT3 (Organic Cation Transporters, SLC22A3; OMIM 604842). PMAT is a 530-aminoacid protein which contains 11 transmembrane segments, whereas OCT3 codifies a 556-aminoacid protein with 12 transmembrane segments. Once inside the enterocyte, metformin enters interstitial fluid and subsequently reaches the liver via the OCT1 (SLC22A1; OMIM 602607), OCT2 (SLC22A2; OMIM 602608) OCT3 transporters, which are located in the basolateral cell membranes [38]. In the liver, the Multidrug and Toxicity Extrusion protein, MATE1 (SLC47A1; OMIM 609832), allows transport of metformin through canaliculi [39]. Once in the circulatory system, metformin enters the kidney via OCT2 which uptakes the drug from blood; and then excretes it through tubular ducts, via OCT1, MATE1 and MATE2 (SLC47A2; OMIM 609833) [40-43]. Despite this wide array of transporters implicated in the distribution and excretion of metformin, only polymorphisms of OCT1 appear to effectively impact the potential pharmaceutical effect of the drug [44,45].
Once inside target cells, metformin induces the activation of anti-tumoral effects through both AMPK-independent and dependent pathways. The former mechanisms include inhibition of the mitochondrial respiratory chain [46], modulation of stemness profile [47], and copper sequestration [48]. The latter property depends on metformin’s chemical properties: As a cationic protein, it is capable of forming a pseudoaromatic square planar complex between 2 dimethylbiguanide and 1 copper ion via delocalization of their π-electron [48,49], a quality which is mandatory for effective anti-hyperglycemic biguanides Figure 2 [50].
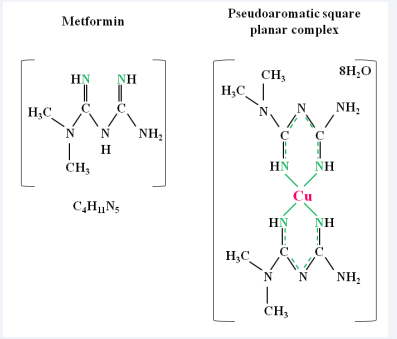
Figure 2: Diagram depicting chemical structure of Metformin and its association with copper
This modification allows the conformation of a pseudoaromatic structure, relying on the van der Waals isosurfaces which make the Y-aromaticity possible [51] and the assembly of Shift-base ligands (Salen-like complexes) [52], interfering with copper mitochondrial metabolism [53]. And even more interestingly, the activation of AMPK is blunted by the absence of the delocated electron, rather than by the phosphorylation of its downstream target, protein S6 [48]. Metformin’s effects on the respiratory chain and stemness profile modulation will be further discussed in the Mitochondrial Toxicity section.
OVERVIEW OF CANCER CELL METABOLISM
The key to understanding metformin as a potential anti-tumoral drug lies in the unique metabolism of cancer cells, which is characterized by a high proliferation rate, increased glucose oxidation, and the capacity to thrive during hypoxia [54]. Two hypotheses –the Warburg Effect and the Crabtree Effect– attempt to explain the acquisition of multiplicative capacity in the ever-increasing hypoxic environment typical of these cells [55,56].
The Warburg effect, named after the iconic publication by Dr. Otto Warburg in 1956 [57], describes the activation of an aerobic glycolytic phenotype, with induction of a “fermentative” metabolism and lactate production (Figure 3).
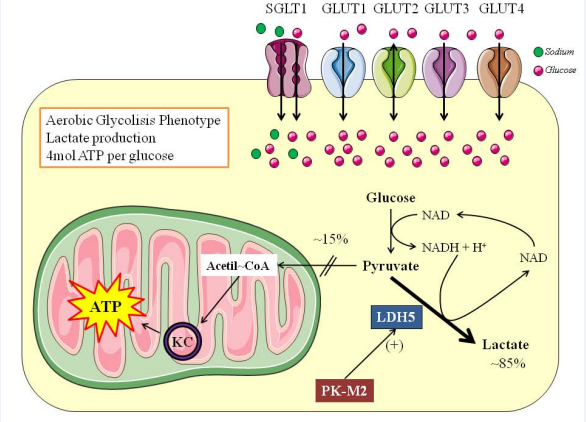
Figure 3: The Warburg effect. This drawing shows the basic changes that occur within a cell which has acquired the aerobic glycolysis phenotype
This scenario is characterized by increased expression of glucose transporters [58], LDH5 isoform [59] and pyruvate kinase M2 (PK-M2) isoform [60], alongside induction of glutaminolysis and acceleration of the Krebs cycle [61]. Because cancer cells survive on a pseudoanaerobic metabolism, glucose uptake must increase parallel to the their needs; therefore, glucose transporters such as SGLT-1 (SLC5A1; OMIM 182380), Glut1 (SLC2A1; OMIM 138140), Glut2 (SLC2A2; OMIM 138160) and Glut3 (SLC2A3; OMIM 138170) are overexpressed in cancers such as colorectal, head and neck, lung, pancreas, endometrial, breast, ovary, and liver [62], while Glut4 (SLC2A4; OMIM 138190) has been observed in breast and gastric cancer [62]. The expression of LDH5 favors the metabolism from pyruvate to lactate due to its higher affinity for pyruvate [63], granting the fermentative phenotype. Finally, the PK-M2 isoform has been considered fundamental to maintain tumor growth and expansion due to its lower affinity for phosphoenolpyruvate (PEP) and lower enzymatic activity [64,65]. However, this lower activity of PK-M2 has not been associated with impaired conversion of PEP to pyruvate; instead, Vander Heiden [66,67] have observed that Phosphoglycerate Mutase-1 (PGM1) can convert PEP by transference of the phosphate to the catalytic pocket of the mutase, concomitantly increasing serine and glycine biosynthesis [66,67].
Aminoacid metabolism in cancer cells is complex, fundamentally encompassing 3 amino acids: glutamine, glycine and serine. Glutaminolysis is key for providing carbon skeletons to fuel the Krebs cycle via production of α-keto-glutarate, increasing the production of ATP [68,69]. Serine de novo synthesis is associated with the increased offer of one-carbons for DNA synthesis and proliferation [70] and acquisition of selective advantage [71], mechanism especially observed in triple negative breast cancer [72]. Interestingly, serine is a known allosteric activator of PK-M2, fulfilling a full circle on the relationship between PK-M2, PGM1, and pyruvate diversion towards mitochondria [66,67,73]. Finally, glycine de novo biosynthesis from threonine is associated with nucleotide synthesis, tumor cell multiplication and poor prognosis [74].
To summarize, it has been confirmed that cancer cells survive on glucose-dependent pathways, whose metabolites are deviated towards the production of lactic acid and ATP. Despite anaplerotic pathways contributing to the Krebs cycle (e.g., glutaminolysis), this process is weakened because it is also used as a source for macromolecular synthesis during cell proliferation [75]. Glucose-induced inhibition of mitochondrial respiration is known as the Crabtree effect [76,77]; Figure 4.
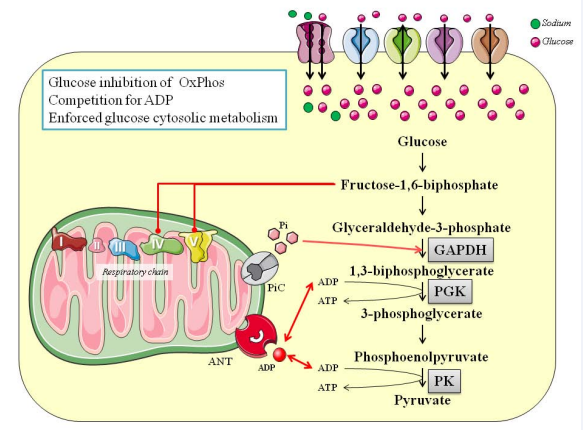
Figure 4: The Crabtree effect. This drawing shows the basic changes that occur within a cell which in the presence of high concentrations of intracellular glucose, develops oxidative phosphorylation inhibition and obligated citosolic ATP production. ANT: Adenosine nucleotide translocator; GAPDH: Glyceraldehyde-3-phosphate Dehidrogenase; PGK; Phosphoglycerate Kinase; PiC: phosphate exchanger; PK: Pyrivate Kinase.
This phenomenon has been related to the competition between glycolysis and respiratory chain for ADP as a main substrate and the effects of calcium inside the mitochondria [77,78]. This “custodial fight” stops mitochondrial functionality, and ATP production becomes exclusively cytosolic [77].
One cell model to explain the intricacy of these effects was published by Suchorolski [79], using Barrett´s esophagus (BE) cell lines. These investigators chose this cell line due to their energetic characteristics during oncogenesis, where esophageal adenocarcinomas show the Warburg effect [80]. Consequently, BE cells are at a crossroads towards choosing a glycolytic pathway or adaptation towards dysfunctional mitochondria. The study [79] demonstrated that in the early stage of BE the cells used oxidative phosphorylation as the main source of ATP, conveying functional mitochondria. Nonetheless, as transformation continues, late stages of BE are mainly Crabtree-dependent yet retain active mitochondria. Finally, when adenocarcinoma fully develops, mitochondrial metabolism is uncoupled and the Warburg effect ensues. In metaplastic cells, the Crabtree effect provides a survival advantage and allows endurance of the tumor cells even during variations of glucose and oxygen availability. These biochemical features are also observed in cancerous cells, such as leukemia cells [81] which demonstrate various metabolic profiles relating to aerobic glycolysis and mitochondrial production of ATP.
METFORMIN AND MITOCHONDRIAL TOXICITY IN CANCER CELLS
As can be observed from the previous sections, viable and active mitochondria are necessary; albeit progressively losing their efficacy and coupling properties during tumor cell expansion and intensification of hypoxia. Mitochondrial toxicity is one of the most effective mechanisms at killing highly replicating cells, such as cancerous cells. Metformin has been proven to exert 2 specific mitochondriotoxic effects: inhibition of respiratory complex I and inhibition of respiratory complex IV and other copper-dependent proteins.
The mitochondrial respiratory chain is a supercomplex ensemble, conformed by Complexes I through IV [82,83]; Figure 5.
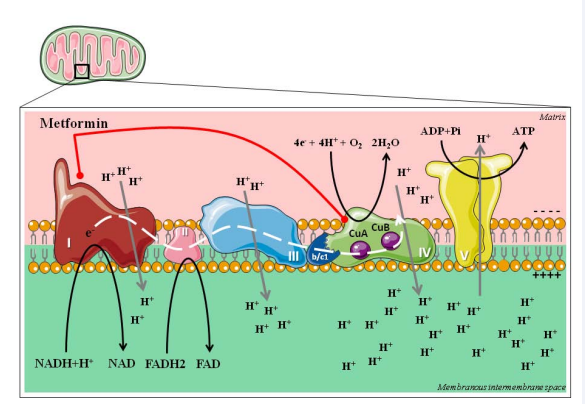
Figure 5: The Respiratory Chain complexes. The respiratory chain (complexes I – IV) are coupled with oxidative phosphorylation (Complex V), due to the need to maintain an electrochemical difference between the mitochondrial matrix and intermembrane space. Complexes I, III and IV and proton pumps which maintain the intermembrane space acidified. Complex V has an ion channel which is used to transport protons towards the matrix and provide voltage necessary for ATP production.
Complex I encompasses an L-shaped protein NADH:ubiquinone oxidoreductase, coupled with Flavin Mononucleotide and 7 Ferrous-Sulfur (FeS) clusters [82-84]. This first complex oxidizes intermediary metabolism-derived NADH to NAD+ , pumping protons towards the intermembrane space and passing electrons towards Complex II. Succinate:ubiquinone oxidoreductase is the enzyme component of Complex II, along with 3 FeS clusters [82,83,85]. This second complex is not a proton pump, yet it serves to mobilize reducing equivalents from FAD2 towards the next complex. The next in line is Complex III, the Cytochrome c oxidoreductase, which oxidizes ubiquinone moving electrons via cytochrome b/c1 towards Complex IV, while pumping protons outside mitochondrial matrix [82,83,86]. Finally, the Cytochrome c oxidase is the last piece of this machinery, ending the reduction of 1 oxygen molecule with 4 protons, rendering 2 H2 O molecules [82,83]. The model of activation and coordinated functioning for respiratory complexes are encircled in the super complexes models [82], where the assembly of all the complexes nearby the ATP Synthase (Complex V) [87] ensures electron flux, controlled reactive oxygen species (ROS) production, and maintains a thermodynamically working environment. The uncoupling of these supercomplexes, especially the separation of Complex I from the rest of the components, is associated with higher generation of ROS [88].
For over 10 years, it has been known that metformin can exert anti-hyperglycemic effects through the inhibition of mitochondrial energy metabolism. Owen [46] designed an elegant in vitro model with hepatoma cells for the evaluation of the inhibitory effect of metformin on mitochondrial membrane potential, reporting that the drug was capable of inducing a time-and dose-dependent blockage of the Krebs cycle, increasing the production of lactate and lactate shuttles. Metformin accumulation on mitochondria depends on a steady membrane potential of ~180 mV, with an average mitochondrial quantum 1000-fold that of plasma concentration (~100 mM); yet the liver it’s the first-passage organ and the levels that circulate in this organ are more than enough to induce this inhibitory effect on the respiratory chain, blocking 50-80% of glycolysis and paralleled induction of glucose uptake via Glut1/4. These results have also been observed in other cells lines such as pancreatic ductal adenocarcinoma [89,90], colorectal cancer [91], and other hepatoma/liver mitochondria experiments [92,93].
Inhibition of Complex I am associated with enhanced production of ROS and induction of apoptosis, observed not only in physiologic mitochondrial investigation [88], but also during experiments with Delocalized Lipophilic Cations (DLC). These cationic proteins are highly hydrophobic positively charged components, intensely drawn towards the mitochondrial matrix, which is negatively charged. It has been recognized that DLC´s inhibit oxidative phosphorylation and cell death, making them perfect weapons against tumor cells [94,95].One of these DLC is [Cu(isaepy)2], also known as Isatin-Schiff base Copper(II), which is a known apoptosis inducer due to respiratory Complex I inhibition in neuroblastoma cells [96]. Induction of cell death was blunted when a copper chelator was incubated with the cells, proving the key role of this ion in this kind of DLC [93]. The chemical structure of [Cu(isaepy)2] is quite similar to that of the pseudoaromatic square planar complex between cupper and metformin [48-53], and they share physical characteristics, which makes it very plausible that metformin works as a DLC molecule when sequestering copper inside the mitochondria, inducing oxidative stress [48,53].
Indeed, copper mitochondrial metabolism is a highly conserved mechanism which relies on copper chaperones and their copper-mobilizing capacity from cytosol to mitochondrial matrix [97,98]. In tumor cells, copper is essential for growth, regulation of oxidative phosphorylation and modulation of glucose expenditure for energy production (99); therefore, copper insufficiency is considered cytotoxic for such cells [99,100]. This copper chelating property of metformin is likely responsible for the inhibition of Complex IV, due to the disassembly of copper binding sites CuA and CuB [101,102]. The Cytochrome c oxidase is fundamental to maintain membrane voltage during high-input periods, thanks to the gating property of Glu242 which is close to Heme α3 and CuB [101].
Overall, metformin seems to be a bona fide copper chelator and DLC, which disrupts activity of Complexes I and IV; and probably interferes with copper-dependent proteins such as Superoxide Dismutase.
THE ROLE OF METFORMIN IN CELL CYCLE AND PLURIPOTENCY MODULATION
As a well-known activator of AMPK [6], the analysis of metformin’s AMPK-independent functions can be quite challenging, especially concerning its effects on the cell cycle. Metformin-activated AMPK is known to consequently activate TSC2 (Tuberous Sclerosis Complex), which inhibits RagC/mTOR1 in several cancer lines from breast cancer [103], pancreatic intreaepithelial neoplasia [104], head/neck [105] and skin [106] squamous cell carcinoma and nasopharyngeal carcinoma [107]. The blocking on mTOR1 modulates processes further down such as: protein synthesis, lipogenesis, pentose phosphate pathway and the very important inhibition of autophagy [108]. However, some properties have been attributed to mechanisms still unclear, yet seemingly AMPK-dependent, such as the control of pluripotency [47] and activation of p53 [109]; Figure 6.
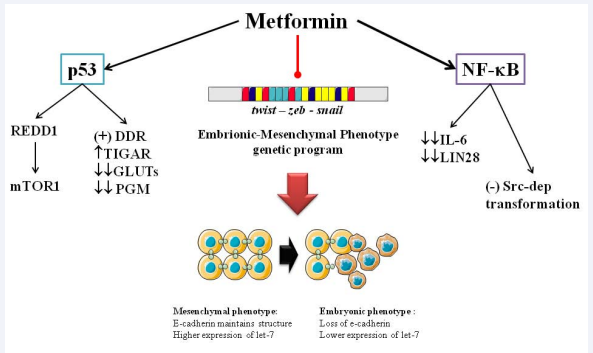
Figure 6: Metformin´s AMPK-independent modulation of cancer: Involving modulation of protein synthesis, inflammation and stemness genetic program. DDR: DNA Damage response; PGM; Phosphoglycerate Mutase-1; REDD: Regulated in Development and DNA Damage responses; TIGAR: TP53-inducible glycolysis and apoptosis regulator.
Vazquez-Martin [110] published their results about the influence of AMPK in the modulation of stemness in induced Pluripotent Stem Cells (iPSC), centering in the expression of reprogramming factors such as Oct4, Klf4, Sox2 and c-Myc. This pioneer work showed that metformin activated-AMPK prevented the expression of Oct4, putting a stop in the “immortalization” process of iPSCs by blocking bioenergetic glycolytic metabotype. Moreover, the same team has reported that metformin is able to modulate Embrionic-Mesenchymal Phenotype (EMP) via restriction of its genetic controllers, twist-zeb-snail and upregulation of microRNA let-7 [111], forcing a mesenchymal phenotype which is a “mature”-like cell, in an AMPK-independent manner. Such results have been also reported in a pancreatic cell lines by Li [112], who also found the increased expression of microRNA-26a and microRNA-192. Likewise, metformin blocks TGF-β activation of the EMP genetic program and with it, loss of expression of E-cadherin in breast cancer cells [113], limiting their migration and metastasis [114]. And finally, this biguanide also intervenes in inflammation modulation, another link between diabetes and cancer [18,19]. Hirsch [115] published their results on the effect of metformin on cancer stem cells, showing that the biguanide inhibits nuclear transcription factor NF-κB via phosphorylation and activation of IKB. The blockage of NF-κB lowers the expression of IL-6 and LIN28 (natural repressor of let-7), and Src-dependent cancerous transformation [115,116].
On the other hand, metformin appears to activate p53, the guardian of the genome [109]. This tumor suppressor-related protein is responsible for one of the most complex intracellular signaling cascades, which not only controls DNA damage response and tumor suppression [117], but is also a glucose metabolism controller [118,119]. Activation of p53 is associated with downregulation of Glut-1/3/4, phosphoglycerate mutase, while upregulation of TIGAR (TP53-inducible glycolysis and apoptosis regulator) and glutamine synthase [115,116]. Using mouse embryo fibroblasts, He [109], determined that AMPK could phosphorylate and inactivate MDMX, a natural repressor of p53, allowing its stabilization and activation. Such property has been related to induction of senescence in endothelial cells [120] and non-small cell lung cancer [121]; and with increased radiation response in melanoma [122] and colon cancer cells [123].
A novel molecular axis has been proposed for metformin, the p53/REDD1 pathway. The Regulated in Development and DNA Damage responses-1 (REDD1) is a hypoxia-related target gene which is associated with stressed cell survival [124], whose main effect is the inhibition of mTOR1 via TSC2 activation [125]. Ben Sahra [126] published new insights on this new pathway, proving that metformin inhibited mTOR1 by inducing p53 and REDD1 in prostate cancer cells, results which have confirmed by Mohammed [104]. As it seems, metformin is able to modulate cancer cell nutrition and fuel-regulated survival [90,104,108], which could impaired proliferation and migration phenotypes.
THE ROLE OF VITAMIN B12 IN METFORMIN´S ANTITUMORAL ACTIVITY
Perhaps the most unusual anti-tumoral effect that has been linked to metformin is its role in the metabolism of monocarbons derived from serine, glycine and threonine. DNA synthesis requires the methyl donation from S-adenosylmethionine (SAM), a unique molecular species generate only on the one-carbon metabolism [127] (Figure 7).
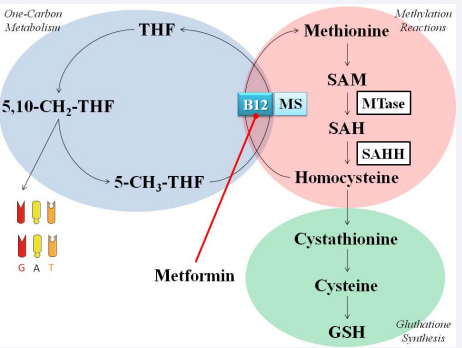
This process requires a numbers of enzymes and it has the property of overlapping the folate, methionine and glutathione cycles [128], requiring the presence of 4 vitamins: folate, vitamine B6, B2 and B12 [129]. The absence of vitamin B12 (cyanocobalamin) compromises the progression of the cycles in a phenomenon called “folate bottleneck”, which is characterized by the blunted production of SAM and stalled DNA synthesis and methylation [128,129].
As previously stated, serine, glycine and glutamine metabolisms are associated with tumor cells proliferation and survival advantage [68-72,74,75]. In fact, inhibition of the one-carbon metabolism using methotrexate, a widely used antifolate chemotherapeutic [130], slows cancer cell proliferation due to halting of the one-carbon metabolism and ATP shortage [131]. Interruption of folate metabolism and stalling of purine synthesis has been proven to induce AMPK and senescence in in vitro models of prostate [132] and breast [133] cancer. Moreover, Vazquez [134] have uncovered novel ATP generation pathways that involve one-carbon metabolism, glycine cleavage and serine biosynthesis, highlighting the importance of this biochemical pathway in cancer cell proliferation, survival, and even, act as a methotrexate-sensitive marker [135].
Cyanocobalamin deficiency has been demonstrated in T2DM patients being treated with merformin [136-138], sometimes associated with megalobastic anemia in this group [139,140]. Bauman [141] suggested that these low levels of B12 were related to decrease absorption due to a calcium-dependent ileal membrane antagonism, and such effect seems amplified with the concomitant use of proton pumps inhibitors [142]. Taking into account that Vitamin B12 deficiency induces a folate bottleneck, a cancer cell which is living in a pseudo-folate deficiency state is even more susceptible to the cytostatic effects of metformin [143]. Therefore, it has been suggested that metformin and methotrexate should be considered as partners in treatment of diseases such as cancer [143] and psoriasis [144].
PERSPECTIVE
Metronomic chemotherapy is a modality of cancer treatment characterized by continuous low-dose administration of conventional chemotherapy drugs, without extended drug-free periods of time [145]. Such therapy has been known to offer better angiogenic control and tumor regression than conventional high-dose schemes [146-148]. This kind of therapy has been known to offer higher survival rates, complete positive responses and fewer cases of grade 3/4 adverse effects [149]. Metformin has been proposed as a novel metronomic chemotherapy drug due to its AMPK-dependent properties [149], especially after the results from Obajimi [132] which evaluated the anti-proliferative effects of AMPK agonists. Nevertheless, as explained in this review, the impact of AMPK-independent effects may be enough to strongly consider the use of metformin as a metronomic agent, especially due to its effects in energy production. However, prospective studies are needed to properly ascertain adequate dosage and time-windows of opportunity; as well as in vitro models in order to fully describe the intracellular effects of metformin.
ACKNOWLEDGMENT
This work was supported by Research Grant no. CC-0437-10- 21-09-10 from Consejo de Desarrollo Científico, Humanístico y Tecnológico (CONDES), University of Zulia, and Research Grant No. FZ-0058-2007 from Fundacite-Zulia.
REFERENCES
31. Gottlieb B, Auld WH. Metformin in treatment of diabetes mellitus. Br Med J. 1962; 1: 680-682.
34. Scheen AJ. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 1996; 30: 359-371.
55. Horsman MR. Measurement of tumor oxygenation. Int J Radiat Oncol Biol Phys. 1998; 42: 701-704.
57. Warburg O. On the origin of cancer cells. Science. 1956; 123: 309-314.
61. Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab (Lond). 2010; 7: 7.
76. Crabtree HG. Observations on the carbohydrate metabolism of tumours. Biochem J. 1929; 23: 536-545.
77. Wojtczak L. The Crabtree effect: a new look at the old problem. Acta Biochim Pol. 1996; 43: 361-368.
114. Zheng H, Kang Y. Multilayer control of the EMT master regulators. Oncogene 2013.
116. Anastasiou D. Metformin: a case of divide and conquer. Breast Cancer Res. 2013; 15: 306.
118. Soga T. Cancer metabolism: key players in metabolic reprogramming. Cancer Sci. 2013; 104: 275-281.
119. Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009; 9: 691-700.
127. Selhub J. Homocysteine metabolism. Annu Rev Nutr. 1999; 19: 217- 246.









































































































































