The First Case Report of A Patient with Coexisting Acromegaly with Adrenocortical Cushing and Pheochromocytoma
- 1. Departments of Endocrinology and Metabolism, Pamukkale University, School of Medicine, Denizli, Turkey
- 2. Department of Pathology, Pamukkale University, School of Medicine, Denizli, Turkey
- 3. Department of Radiology, Pamukkale University, School of Medicine, Denizli, Turkey
Abstract
Pheochromocytoma [PHEO] and Cushing syndrome [CS] may sometimes coexist and very rarely originate from the same adrenal gland. There were 12 cases in the literature in which corticomedullary involvement has been reported. A few cases of primary aldosteronism coexisting with Pheochromocytomahave been reported so far. Also there is very little information about pheochromocytoma associated with Cushing’s syndrome. Cushing’s syndrome secondary to ectopic adrenocorticotropic hormone [ACTH] secretion has been frequently reported nowadays. Our literature search revealed no previous reports of a patient with PHEO and CS originating from the same adrenal gland and coexistent with acromegaly
Keywords
• Acromegaly
• Cortical adenoma
• Cushing Syndrome
• Pheochromocytoma
Citation
Fenkci S, Yalcin N, Sagdas E, Topsakal S, Yaylali GF (2019) The First Case Report of A Patient with Coexisting Acromegaly with Adrenocortical Cushing and Pheochromocytoma. J Endocrinol Diabetes Obes 7(1): 1119.
INTRODUCTION
Adrenal lesion composed of a mixture of different cell types derived from both pheochromocytes and cortical cells, which do not share a common embryological origin [1]. Approximately, 15 cases have been reported in various literatures, since mixed corticomedullary adrenal tumors [MCMT] was first described in 1969 [1].Coincidental coexistence of adrenal cortical adenomas and pheochromocytomas,especially in the same adrenal gland, is extremely rare, that’s why it is not considered a category of MCMT. In all of these cases, there were no typical pheochromocytoma symptoms, and the disease was diagnosed by biochemical laboratory results and imaging studies [2]. Interestingly, our patient presented with typical Cushingoid features and had compatible CT imaging suggestive of an adrenal cortical adenoma. The 24-hour urine metanephrine, normetanephrine, and catecholamines were normal, but preoperatively her blood pressure was not under control.
CASE PRESENTATION
A 32-year-old woman was referred to our clinic due to diabetic ketoacidosis. Her past medical history revealed that she had been treated with levothyroxine for Hashimoto’s thyroiditis. At physical examination, marfanoid appearance and plethora were noted, and her blood pressure was 190/140 mmHg. She had common signs and symptoms of Cushing syndrome involving truncal obesity [weight gain of nearly 12kg mainly around the midsection and upper back], buffalo hump and moon face over the past 14 months, without evidence of irregular or absent menstrual periods,easy bruising, purple striae, acne and hirsutism. BMI was calculated as 26.95kg/m2 .
The baseline C peptide level was 2.4 ng/mL, anti-gad and antiinsulin antibodies were negative, therefore she was considered as type 2 diabetes.Intravenous insulin and fluid therapy were started and then switched to basal-bolus insulin therapy. During follow-up, blood glucose regulation was achieved with basal– bolus insulin therapy, but blood pressure wasn’t under control despite of antihypertensive drugs. Thus, the blood pressure was reduced to 160/100mmHg with combined antihypertensive therapy. Hence, secondary causes of hypertension were investigated.
Laboratory examinations were summarized in table 1.
Table 1: Laboratory examinations of the patient.
| Parameters | Level | |
| Cortisol | ||
| Baseline [µg/dL] | 16.00 | |
| After 2mg dexamethasone suppression test [µg/dL] | 14.10 | |
| Midnight [µg/dL] | 17.60 | |
| After 8mg dexamethasone suppression test [µg/dL] | 14.20 | |
| Urinary free [µg/day] | 232.71 | |
| Metanephrine [normal range: 0-390mg/day] | 110.22 | |
| Normetanephrine[normal range: 0-320mg/day] | 169.53 | |
| Vanilmandelic acid[normal range: 3.3-5.5mg/day] | 3.72 | |
| Growth hormone [ng/mL] | 22.20 | |
| Aldosterone [ng/dL] | 18.20 | |
| Renin [ng/mL/hr] | 2.65 | |
| Prolactin [ng/mL] | 45.41 | |
| Ca++[mg/dL] | 9.49 | |
| TSH [uIU/mL] | 4.26 | |
| TSH: thyroid-stimulating hormone | ||
OGTT/ growth hormone [GH] suppression test showed that GH responses were as 22.3; 18.3; 22.4; 19.6ng/mL, respectively. Renal Ultra sonography and Doppler was in normal range. Screening tests for etiology were done. In pituitary MRI, the macro adenoma with less contrast enhancement was detected in the post-contrast series compared to the 16x15x17mm normal pituitary parenchyma, which showed sphenoid sinus extension in the inferior part of the adenohypophysis. Well-contoured, well-defined, 35x23 mm sized adrenal suppressed adenomas were detected in the left adrenal gland by adrenal magnetic resonance imaging [MRI] [Figure 1].
![Adrenal magnetic resonance imaging [MRI] left adrenal mass.](https://www.jscimedcentral.com/public/assets/images/uploads/image-1705735864-1.png)
Figure 1 Adrenal magnetic resonance imaging [MRI] left adrenal mass.
Appearance suggested that the adenoma had a dense cellular characteristic. In the light of the aforementioned data, our diagnosis for the patient was Cushing syndrome accompanied with acromegaly.
Surgery was performed for adrenal adenoma as first step treatment. Resected left adrenal tissue weighed 30gr and measured as 6x5x3cm sizes. Macroscopically, the tumor excised was a 4x3x2cm encapsulated mass, which consisted of two parts: i] dark brown Pheochromocytoma and ii] yellowishorange adrenal cortical adenoma [Figure 2].
![Macroscopically, the tumor excised was a 4x3x2cm encapsulated mass, which consisted of two parts: i] dark brown Pheochromocytoma and ii] yellowish-orange adrenal cortical adenoma.](https://www.jscimedcentral.com/public/assets/images/uploads/image-1705735877-1.png )
Figure 2 Macroscopically, the tumor excised was a 4x3x2cm encapsulated mass, which consisted of two parts: i] dark brown Pheochromocytoma and ii] yellowish-orange adrenal cortical adenoma.
On the microscopic evaluation of the tumor tissue, it was observed that two tumors were localized side by side in the adrenal cortical adenoma and pheochromocytoma in the tumor tissue. Histopathological examination did not reveal mitotic activity and vascular invasion. Also, capsule and surrounding connective tissue were intact. In the immune histochemical staining, focal +, Melan-A +, synaptophysin +, chromogranin A +, Ki-67 1% were detected [Figure 3].
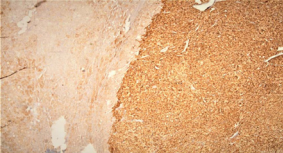
Figure 3 Histopathological examination of the adrenomedullary adenoma.
Post-operatively, adrenal insufficiency ensued, which necessitated continuing hydrocortisone replacement. Following the stabilization, the patient was discharged from the hospital on hydrocortisone 20 mg per day and sandostatin LAR/month. Transsphenoidal pituitary adenectomy was performed 3 months later. Immuno histochemical study showed that pituitary adenoma positively stained with GH. Sandostatin LAR treatment was continued for residual diseases after the surgery. Steroid and L-thyroxine replacement were already given. Finally, the patient was negative for RET [RE arranged during Transfection] protooncogene.
Post-operative 75-g oral glucose tolerance test [OGTT] was performed. The results of the 75-g OGTTs from after surgery revealed a partially postoperative improvement [0 min: 92mg/ dL, 60 min: 176mg/dL, 120 min: 221mg/dL respectively]. Metformin treatment was commenced. Her preprandial glucose levels with metformin detected as 105 mg/dL and postprandial as 126 mg/dL and HbA1c level as 6.5%. Also, in the postoperative period, hypertension of the patient was disappeared. Her mean blood pressure was 125/80mmHg.
DISCUSSION
Pheochromocytoma [PHEO] and Cushing syndrome [CS] may sometimes coexist and very rarely originate from the same adrenal gland. There have been 12 cases in the literature in which corticomedullary involvement has been reported. Very rarely, however, over secretion of both adrenomedullary and adrenocortical hormones from the same adrenal gland has been reported. A few cases of primary aldosteronism coexisting with Pheochromocytoma have been reported [3].
There is very little information about pheochromocytoma associated with Cushing’s syndrome. Recent studies on literature reporting Cushing’s syndrome cases depending upon ectopic ACTH production. In a review paper, mentioned 24 case reports of ectopic Cushing’s syndrome. Most of these cases had clear Cushing’s syndrome or pheochromocytoma clinics, and only two of them did not show clinical manifestations of excess catecholamine. In our presented patient serum methanephrine and WMA levels were found to be within normal limits, despite the presence of resistant hypertension. But in literature this was the first patient with PHEO and CS originate from the same adrenal and acromegaly [4-6].
In such cases, the pituitary gland may appear enlarged but in our patient there was a macro adenoma in the pituitary gland. Diabetes mellitus occurs in nearly 10% of patients with acromegaly and is secondary to insulin resistance caused by high levels of growth hormone. Diabetes ketoacidosis has been described as a rare complication of acromegaly, resulting from a relative insulin deficiency caused by growth hormone excess. This case shows the rare association between diabetic ketoacidosis and acromegaly [7-9]. Catecholamine excess additionally induced ketoacidosis.
An increased risk of certain types of cancers, including colorectal, breast, prostate and thyroid cancers, has been previously described in patients with acromegaly [10]. However, little is currently known about the association between acromegaly and adrenal tumors. One case-control study by Scaroni et al. found that adrenal adenomas/hyperplasia were more frequently observed in acromegalic patients [11] than in the general population and that the baseline endocrine data for the patients with adrenal masses showed no hormone hypersecretion.
Previous clinical studies assessing the associations between adrenal gland tumors and acromegaly have reported correlations between a larger tumor size [12] and an increased prevalence of adenoma or hyperplasia [13] as well as acromegaly. However, it remains controversial whether there is a causal relationship between adrenal morphological alterations and the disease activity. In conclusion, we herein reported a rare case of the PHEO and CS originating from the same adrenal gland and coexistent acromegaly.
The patient did not fulfill the criteria of the Carney complex Carney complex is a familial lentiginosis syndrome; these disorders cover a wide phenotypic spectrum ranging from a benign inherited predisposition to develop cutaneous spots not associated with systemic disease to associations with several syndromes. In carney complex the adrenocortical disease presented with primary pigmented nodular bilateral hyperplasia [14] Spotty skin pigmentation which is the most clinical presentation of the syndrome was not found in our patient. A novel MEN syndrome was discovered, initially in rats where it was named ‘MENX’ and then in humans, now known as MEN4. Recently, somatic or germline mutations in CDKN1B were also identified in patients with sporadic PHPT, small intestinal neuroendocrine tumors, lymphoma and breast cancer, demonstrating a novel role for CDKN1B as a tumor susceptibility gene for endocrine and other neoplasms [15]. Since the identification of mutations in CDKN1B as causative for MEN4, only 19 cases have been reported in the medical literature. However, no guidelines exist, patients with asymptomatic or symptomatic PHPT that are young [typically <30 years old], with multigland disease, parathyroid carcinoma or atypical adenoma, or those with a family history or evidence of syndromic disease and negative for MEN1 or RET, genetic testing for CDKN1B should be pursued. As we already demonstrated that our patient negative for ret proto-oncogene. Our case did not meet the criteria of our patient’s MEN syndromes. This is the first case in the literature PHEO with CS coexist in the same adrenal with acromegaly.
Cell replication errors may lead to numerical changes in chromosomes, chromosomal translocations, amplification and/ or loss of genes, somatic sequence alterations in specific genes including DNA repair genes, and other genomic changes. This presented patient may be represents a new another candidate genes for multiple endocrinopathies.









































































































































