Transcription Factor Gli-Similar 3 (Glis3): Implications for the Development of Congenital Hypothyroidism
- 1. Division of Intramural Research, National Institute of Environmental Health Sciences, USA
Abstract
Congenital hypothyroidism (CH) is the most frequent endocrine disorder in neonates. While several genetic mutations have been identified that result in developmental defects of the thyroid gland or thyroid hormone synthesis, genetic factors have yet to be identified in many CH patients along with the mechanisms underlying their pathophysiology. Mutations in the gene encoding the Krüppel-like transcription factor, GLI-similar 3 (GLIS3) have been associated with the development of a syndrome characterized by congenital hypothyroidism and neonatal diabetes and similar phenotypes were observed in mouse knockout models of Glis3. Patients with GLIS3-mediated CH exhibit diminished serum levels of thyroxine (T4) and triiodothyronine (T3) and elevated thyroid stimulating hormone (TSH) and thyroglobulin (TG). However, the inconsistent presentation of clinical features associated with this CH has made it difficult to ascertain a causative mechanism. Future elucidation of the biological functions of GLIS3 in the thyroid will be crucial to the discovery of new therapeutic opportunities for the treatment of CH.
Keywords
• Congenital hypothyroidism
• Neonatal diabetes
• Gli-similar 3
Citation
Lichti-Kaiser K, ZeRuth G, Jetten AM (2014) Transcription Factor Gli-Similar 3 (Glis3): Implications for the Development of Congenital Hypothyroidism. J Endocrinol Diabetes Obes 2(2): 1024.
ABBREVIATIONS
CH: Congenital Hypothyroidism; GLIS3: Gli-similar 3; T4: Thyroxine; T3: Triiodothyronine; TH: Thyroid Hormone; TRH: Thyrotropin Releasing Hormone; TSH: Thyroid Stimulating Hormone; TG: Thyroglobulin
INTRODUCTION
The thyroid, through the production of thyroid hormone (TH) by the follicular cells, is essential for normal development, growth, and metabolism of essentially all human tissues [1]. The thyroid gland secretes predominately T4 that is converted into the active form T3 by the intracellular iodothyronine deiodinases in peripheral tissues. After transport and activation in the cell, T3 can interact with nuclear TH receptors and activate or inactivate the transcription of TH responsive genes. The production of TH in the thyroid gland is regulated by the hypothalamus-pituitary-thyroid axis negative feedback loop. For example, low levels of serum TH result in increased release of TSH from the anterior pituitary. TSH itself is regulated by thyrotropin releasing hormone (TRH) produced by thehypothalamus. TSH stimulates the synthesis and secretion of TH in the thyroid to restore circulating hormone levels [2-4]. As part of a negative feedback loop, TH negatively regulates the release of TSH and on the activity of TRH.
The most common hereditary endocrine disorder, CH, is defined by sub-physiological levels of thyroid hormoneand most commonly due to defects in thyroid organ development or thyroid hormone synthesis. A number of genetic mutations have been identified that result in developmental defects of the thyroid gland (NKX2-1, NKX2-5, TSHR, PAX8,FOXE1) or defective thyroid hormone synthesis (SLC5A5, SLC26A4, TPO, DUOX2, DUOXA2, GNAS, IYD); however, the genes defective in many patients with congenital hypothyroidism have yet to be identified along with the molecular mechanisms underlying their pathophysiology [5- 7]. Mutations in the gene encoding the Krüppel-like transcription factor, GLIS3 have been associated with a rare syndrome (NDH) characterized by congenital hypothyroidism and neonatal diabetes [8,9]. Patients with NDH exhibit diminished levels of T3 and T4 along with elevated TSH and TG. Patients additionally develop hyperglycemia and hypoinsulinemia often accompanied by polycystic kidney disease, hepatic fibrosis, glaucoma, and mild mental retardation depending on the nature of the mutation [8,10]. Similar phenotypes were observed in mouse knockout models of Glis3 [11,12]. In addition, a number of genome-wide association studies have found a link between several polynucleotide polymorphisms (SNPs) in GLIS3 (rs10758593, rs7020673, rs7034200) and an increased risk for the development of both type 1 and type 2 diabetes, while another SNP (rs514716) was linked to the development of Alzheimer’s disease [13-21]. This review aims to briefly summarize what is currently known about Glis3 and its association with hypothyroidism.
Transcription Factor Glis3
The Gli-similar family of Krüppel-like zinc finger proteins is comprised of three proteins, Glis1-3. Glis1 was first identified by a yeast-two-hybrid screening using the ligand-binding domain of the retinoic acid-related orphan receptor γ (RORγ) as bait [22]. Subsequently, two additional members of the family were identified that possessed high levels of homology with the zinc fingers of Glis1 and were termed Glis2 and Glis3 [23-26]. The Glis proteins are evolutionarily conserved across species dating back to fishes, while a Glis homologue referred to as lame duck/ gleeful(lmd/gfl),was identified in Drosophila [27,28]. Similarity between Glis1-3 is limited to their five tandems Cys2 -His2 zinc finger motifs shared be each member. The Glis zinc finger motifs also exhibit a great degree of homology to the zinc finger domains of the Gli and Zic proteins, giving the Glis proteins their namesake [22,24-26].
The human GLIS3 gene is located on chromosome 9p24.2 and encodes a protein that is approximately 90 kD in size. Several alternate transcripts of GLIS3 have been reported, but to date, no evidence has surfaced revealing a physiological function for the splice variants [8]. Glis3 is expressed in a tissue-specific manner with the highest levels of expression observed in the kidney, thyroid gland, endocrine pancreas, thymus, testis, and uterus [8,24]. Lower levels of expression have also been reported in the brain, lung, ovary, and liver. During mouse development, Glis3 is expressed in a spatio-temporal manner being detected as early as E8.0in the node of mice, a precursor of the notochord, as determined by whole mount in situ hybridization [24]. Glis3 continues to be expressed dynamically throughout neurulation and is later expressed in the developing eye, kidney, and testes. In the endocrine pancreas, Glis3 is detected as early as E11.5 with expression increasing at E12.5 during endocrine differentiation [12]. Pancreatic expression of Glis3 continues into maturity where it is limited to the ß cells of the pancreatic islets and the ductal epithelium [8,12].
Additionally, elevated levels of GLIS3 have been identified in several human cancer cell types involving both the kidney and nervous system.GLIS3 has been reported to be up regulated in chromophobe renal cell carcinoma, a relatively rare neoplasm of the kidney [29]. GLIS3 was also found to be overexpressed in the tumors of the central nervous system known as ependymomas that were associated with high rates of proliferation and poor patient prognosis [30]. Finally, increased expression of GLIS3 was also detected in high-grade proneural glioblastomas [31]. How increased expression of GLIS3 affects the progression of cancer and the pathways acted upon are not clear at this time.
Mechanisms of Action: The Glis3 protein consists of the centrally located zinc finger domain (ZFD), a C-terminal transactivation domain (TAD), and a relatively large N-terminus, the function of which is chiefly unknown [24]. The ZFD is primarily responsible for the recognition ofspecific DNAresponse elements. Through use of an in vitro strategy, the optimal Glis3 binding site (GlisBS) was determined to be 5’-(G/C) TGGGGGG (A/C) [32]. Glis3 is additionally capable of interacting with the consensus Gli-binding site (GBS), 5’-GACCACCCA in vitro, albeit with a lower affinity than for the GlisBS [24,32]. This suggests that Glis3 has a high affinity for GC-rich DNA elements. In vivo Glis3 binding to specific DNA sequences is likely influenced bythe promoter context, the recruitment of co-regulators, and possibly post-translational modifications of Glis3. The fact that Glis3 and members of the Gli- and Zic-families can recognize similar binding motifs also allows for the possibility of cross-talk between the Glis and the Gli/Zic signaling pathways [33,34].
In addition to its role in GlisBS binding, the ZFD of Glis3 also plays an important role in determining the subcellular localization of Glis3. Fluorescent-tagged Glis3 has been observed predominately in the nucleus of growing cells [24,35], whereas deletion of Glis3 ZF4 or disruption of its tetrahedral configuration by site-directed in vitro mutagenesis effectively prevented nuclear accumulation of Glis3, while loss of its other ZFs did not have a discernible effect on its localization [32].
Glis3 has additionally been reported to localize to the primary cilium [28,36]. The primary ciliumisa hair-like organelles that extends from the apical surface of nearly all mammalian cells, including the follicular epithelial cells of the thyroid, and serves as a signaling hub for a variety of pathways [37-43]. The primary cilium is immotile, comprised of 9 doublet microtubules (9+0) enclosed within the plasma membrane. Components of the Shh, Wnt, and PDGF signaling pathways have been found to localize within the primary cilium and ciliary localization appears to be required for their proper signaling [41,44-46]. In addition, certain G protein-coupled receptors, including somatostatin, serotonin, and dopamine receptors and the melanin concentrating hormone receptor, localize to the primary cilium [47-50]. Serving as a sensory organelle, the cilia help detect mechanical stress and osmolarity and serve as chemo- and photo sensors for the cell. The precise role of Glis3 in the primary cilia is not yet known although Glis3 does not appear to be essential for proper cilium formation [12,28]. Based largely on the fairly well-characterized role of Gli-proteins in the primary cilium, it is hypothesized that an external signal(s) likely control localization of Glis3 to the primary cilium within which, Glis3 may be converted to an activator or repressor form following post-translational or proteolytic modification [33,34]. Activator or repressor forms of Glis3 would then translocate to the nucleus where they regulate the transcriptional activity of target genes. Future studies will have to be conducted to determine the precise role of the primary cilium in Glis3 signaling but in support of the idea that the cilia play an important in Glis3 signal transduction, aberrant Glis3 signaling is implicated in several pathologies associated with defects in the primary cilia.
Glis3-mediated gene regulation: To date, only Ins2, Ngn3, and Fgf18 have been demonstrated to be direct targets of Glis3 [12, 51-54]. In each case, Glis3 acts as a transcriptional activator through binding to one or more GlisBS within the 5’ upstream regulatory regions of these target genes. Additionally, Glis3 effectively induces the transactivation of a reporter under the control of six tandem copies of the GlisBS. This activation as well as the transactivation of reporters under the control of the mIns2 promoter was dependent on the presence of the Glis3 C-terminal transactivation domain [12,24,35,55]. For reasons that are not clear, deletion of the Glis3 N-terminus up to amino acid 302 increased Glis3-mediated transactivation, while transactivation activity decreased with subsequent deletions [12,35]. These observations indicated that a repressor domain(s) may be located within the Glis3 N-terminus or that N-terminal deletions inducechanges in protein folding thatinfluence interactions with co-regulators and/or DNA.
Gene regulation by transcription factors often requires interactions with co-regulatory proteins. Several proteins that interact with Glis3 have been identified. The tumor suppressor and negative regulator of hedgehog signaling, suppressor of fused (Sufu) has been shown to interact with a VYGHF motif within a 58 amino acid region of the Glis3 N-terminus that shares high levels of homology with a corresponding region found in members of the Gli protein family [35]. Exogenous Sufu expression stabilized Glis3 protein levels in a manner that required interaction between the two proteins and decreased Glis3 polyubiquitination suggesting that interaction protected Glis3 against proteasomal degradation. Additionally, Sufu over-expression repressed Glis3- mediated Ins2 activation, but whether this was due to the action of Sufu as a transcriptional repressor or due to the decreased Glis3 turnover has not yet been resolved.
The transcriptional co-regulator, transcriptional co-activator with PDZ-binding motif (Taz/Wwtr1) was identified as a protein that interacts with a PPXY motif located in the C-terminus of Glis3 [36]. Taz is a component of the Hippo signaling pathway and has been suggested to have roles in controlling cell proliferation, planar-cell polarity, and epithelial-to-mesenchymal transition. Importantly, defects in either Taz or Glis3 promote the development of polycystic kidney disease suggesting a possible link through these two proteins [8,36,56-58].
Finally, Glis3 has been shown to interact with several regulators of insulin transcription, pancreatic duodenal homeobox 1 (Pdx1), neuronal differentiation 1 (NeuroD1), and v-mafmusculoaponeurotic fibrosarcoma oncogene homolog A (MafA) [51]. Glis3 synergistically activates transcription of the Ins2 gene along with these three co-regulators through binding to their respective enhancer elements located within the gene’s proximal upstream promoter region [51,55]. The proteins likely form a regulatory complex through mutual interactions with the ubiquitous co-activator, CBP/p300 and studies have shown that in the absence of Glis3 or the two GlisBS located within the Ins2 promoter, Pdx1 and MafA binding to the insulin regulatory region is severely reduced [55]. These findings suggest that Glis3 may be required to maintain insulin expression in the mature ß cell and is consistent with studies showing that GLIS3 is down regulated in patients with type 2 diabetes mellitus and conditional knockout of Glis3 in the mature pancreas results in the development of hypoinsulinemia and hyperglycemia [59,60]. Further studies are needed to determine the precise role of Glis3 in regulating insulin transcription.
In addition to its role in the maintenance of the mature ß cell, transcriptional regulation mediated by Glis3 is also imperative in the development of the endocrine pancreas. Mice with ubiquitous knock-out of Glis3 only survived for several days after birth and displayed severe hyperglycemia and hypoinsulinemia [12]. In addition to decreased levels of the pancreatic hormones insulin, somatostatin, glucagon, and pancreatic polypeptide, Glis3-null mice also had severe reductions in the levels of ß cell markers such as MafA, Nkx6.1, and Glut2 suggesting a general absence of ß cells. The bHLH transcription factor, Ngn3, which is critical in the endocrine progenitor cells, was also dramatically reduced in Glis3 knock-out mice leading to the conclusion that Glis3 is required for the formation or maintenance of these progenitors. Indeed, recent studies have indicated that Glis3 can directly regulate transcription of Ngn3 through binding to its promoter [52,53].
Congenital hypothyroidism
CH, a heterogeneous condition resulting from decreased or absent action of thyroid hormone,is the most frequent congenital endocrine disorder in neonates with an incidence of 1:2000 to 1:4000 [61]. Clinical features of CH are subtle and non-specific during the neonatal period due in part to the passage of maternal thyroid hormone across the placenta; however, early symptoms may include long-term jaundice, difficulty feeding, lethargy, constipation, macroglossia, hypothermia, edema, wide posterior fontanel, hoarse cry, and umbilical hernia [62]. Clinical newborn screening, based on the measurement of hormone levels, is used routinely to identify hypothyroid infants soon after birth, but if CH remains untreated, the clinical features become more evident after six months of life with growth retardation, delays in motor development, and permanent intellectual disability. Prevention of cretinism and optimal neurological development can be achieved in affected infants by early introduction of hormonal replacement therapy.Levothyroxine is the treatment of choice used to raise serum T4 and normalize serum TSH levels. In general, the prognosis for infants detected by screening and started on treatment early is excellent, with neurocognitive outcomes similar to sibling or classmate controls [63].
Primary CH, the most common form of CH, occurs as a result of abnormal thyroid gland development (thyroid dysgenesis) or disruptions in thyroid hormone biosynthesis (thyroid dyshormonogenesis). Less common causes of CH are secondary or peripheral and result from defects in TSH synthesis or action or in thyroid hormone transport, metabolism, or action [61]. Structural defects of the thyroid gland including athyrosis, ectopic gland, and thyroid hypoplasia, account for the majority of cases of CH (80%). The remaining 20% of children diagnosed with CH are affected by an inborn defect in thyroid hormone synthesis, which occurs in most cases as an autosomal recessive trait of inheritance. Reduced hormone secretion due to thyroid dyshormonogenesis and the resulting diminished negative feedback on the pituitary gland leads to increased TSH secretion stimulating the thyroid gland. As a result, these patients are either born with an enlarged thyroid gland or are susceptible to thyroid hyperplasia postnatally.
Genetic Causes of CH: Insights into the etiology of CH have revealed that genetic causes are detectible not only in patients with dyshormonogenesis, but also those with developmental defects of the thyroid, which were previously thought to be sporadic in occurrence. Only a few cases of familial thyroid dysgenesis have been reported and variance existed even between monozygotic twins [6,64,65]. Nevertheless, studies have shown that 2% of all cases of thyroid dysgenesis were familial in occurrence and that 7.9% of first degreerelatives of infants with CH had a thyroid developmental anomaly [64,66]. Inactivating mutations that contribute to thyroid dysgenesis have been identified in genes that encode transcription factors that are expressed in thyroid embryogenesis and in the normal functioning thyroid gland, including PAX8, FOXE1, NKX2.1, and NKX2.5 [67-75]. These transcription factors are expressed in other tissues in the developing fetus; therefore, their inactivation results in distinct multisystem phenotypes that are linked to their expression. In addition to thyroid dysgenesis, mutations in FOXE1 result in choanal atresia, cleft palate, and spikey hair referred to as Bamforth-Lazarus Syndrome [76]. Mutations in NKX2.1 have been associated with respiratory distress, ataxia, and benign chorea, while mutations in NKX2.5have been linked to cardiac malformations [75,77-79]. PAX8 mutations have been reported to also lead to unilateral kidney agenesis and although PAX8 is also expressed in the brain, no further central nervous system defects have been described [72,80,81]. In addition to transcription factor defects, inactivating mutations in the TSH receptor gene (TSHR) were found to result in CH and thyroid dysgenesis with autosomal recessive inheritance. However, given that TSHR is expressed late in thyroid development, inactivating mutations lead to relatively mild thyroid hypoplasia [82].
Hereditary defects in nearly all of the steps of thyroid hormone biosynthesis have been described and are generally associated with a normally placed thyroid gland and transmitted in an autosomal recessive manner. Considerable progress has been made in the knowledge of mechanisms involved in TH synthesis and release. Briefly, it has been demonstrated that iodide is actively transported by the Na+ /I– symporter (NIS, encoded by SLC5A5) at the thyrocytebasolateral membrane and transported, at least in part, by pendrin (PDS, encoded by SLC26A4), at the apical membrane in to the lumen. Thyroperoxidase (TPO), with hydrogen peroxide generated by dual oxidase 2 and its maturation factor (DUOX2/DUOXA2), catalyzes the one-electron oxidation of iodine andtyrosyl residues, producing monoiodotyrosine (MIT) and diiodotyrosine (DIT) within the thyroglubulin (TG) complex. The same reaction catalyzes the coupling of two iodotyrosine residues to produce T4 and smaller amounts of triiodothyronine (T3). Iodinated TGis hydrolyzed in the lysosomes by cathepsins and TH released from the TG backbone. Finally, released iodotyrosinesare dehalogenated by iodotyrosinedeiodinase (IYD) allowing for the recycling of iodine and tyrosine for further hormone synthesis [7].
Most frequently, dyshormonogenesis is due to defects in TPO activity [83]. CH patients with TPO mutations are afflicted with total or partial iodine organification defects depending on the severity of the mutation, a condition that requires lifelong hormone replacement therapy [83-85]. Inactivating mutations in the SLC5A5 gene have been associated with congenital iodide transport defects, demonstrated by decreased or absent radioactive iodide uptake, and hypothyroidism [86,87]. Inactivating mutations of the SLC26A4 gene are a cause of Pendred Syndrome, characterized by congenital deafness, hypothyroidism and goiter. Defects in pendrin lead to partial iodine organification defects and dyshormonogenesis; however, the hypothyroid phenotype in Pendred Syndrome is typically alleviated with adequate nutritional iodide intake [88-90]. Defects in TG as a cause of CH are most common in Japanese populations, accounting for over one fourth of dyshormonogenesis cases. Iodide organification is not affected given that in the absence of TG, iodide is bound to other proteins such as albumin. Patients with TG defects typically exhibited decreased levels of serum TG and increased levels of serum TSH [91-93]. More recently, mutations in DUOX2 and DUOXA2 have been identified. Although most dyshormonogenesis mutations are autosomal recessive, DUOX2 mutations can be autosomal dominant [94]. Their phenotype is heterogeneous and associated witheither permanent or transient hypothyroidism due to the compensatory activity of DUOX1/ DUOXA1, which are also expressed in thyrocytes, although at a lower level. DUOX2/DUOXA2 mutations are associated with partial iodide organification defects and, as such, can be alleviated with nutritional iodide intake [7,95-97]. Finally, loss of IYD prevents iodide recycling, leading to urinary excretion of MIT and DIT. The resulting iodide deficiency may not be detectable at birth increasing the risk of late diagnosis and treatment [98-100].
Glis3 and CH: Mutations in GLIS3 have been associated with a rare syndrome, NDH, characterized by CH and neonatal diabetes. Patients with NDH exhibit hyperglycemia, hypoinsulinemia, reduced levels of T3 and T4, and elevated levels of TSH and TG.Neonatal diabetes and CH may be accompanied by polycystic kidney disease, hepatic fibrosis, glaucoma, osteopenia, and mild mental retardation depending on the nature of the mutation [8,9]. GLIS3 mutations in humans and knockdown in mice are consistently associated with neonatal diabetes and CH. Glis3- knockout mice die within one week after birth likely due to the severity of neonatal diabetes. As described above, data have suggested that Glis3 plays a critical role in themaintenance or proliferation of endocrine progenitor cells and in the development and maintenance β cells [11,12]. Hypothyroidism was also observed in Glis3 knockout mice; however, histological examination of the thyroid gland suggested that Glis3 does not significantly affect thyroid gland development [11].
Recent evidence has shown that TH also promotes postnatal pancreatic ßcell proliferation and development. In rats, pancreatic islet area and diameter was decreased in neonatal hypothyroid animals compared to the control group [101,102]. Several studies have indicated that T3 has a pro-survival effect on pancreatic isletsin vitro and in vivo. T3 treatment counteracted the onset of Streptozotocin (STZ)-induced diabetes in wild type mice by inhibitingβ-cell death. Moreover, T3 administration prevented the STZ-dependent alterations in serum glucose and insulin levels, islet parameters, TUNEL staining, and the activation of caspases [103]. On the other hand, genetic mouse models with a disruption in the T3-inactivating deiodinase3 (Dio3) gene were found to be glucose intolerant due to impaired islet function. Pancreatic ßcells express high levels of Dio3 and appear to be functionally sensitive to T3 [104]. In pancreatic ßcell lines, T3 treatment has been reported to promote cell proliferation and viability via the regulation of cell cycle-related molecules and to inhibit the apoptotic process via the regulation of pro- and anti-apoptotic factors [105,106]. In addition to cell survival, T3 plays a role in islet function. T3 treatment improved islet function evaluated by insulin secretion in culture by specific activation of Akt [107,108]. T3 also promoted the functional maturation of islets in culture by the induction of ßcell-specific transcription factor MafA [109]. Finally, T3 induced cell cycle perturbations in a pancreatic duct cell line, PANC-1, and played a role in transdifferentiation into insulin secreting β-cell-like cells [110]. While the role of TH in β-cell function and diabetes is becoming clear, the extent to which Glis3-mediated hypothyroidism exacerbates comorbid ßcell defects and diabetes needs further investigation.
Mutations in GLIS3 have been identified in ten patients from seven families with nine of the ten born to consanguineous parents. The nature of the GLIS3 mutations included a frame shift mutation resulting in premature termination, deletion mutations encompassing the 5’ UTR, exons 1-2, 1-4, or 5-9, and a missense mutation (C536W) in the zinc finger motif predicted to affect DNA binding (Figure 1) [8-10,111].
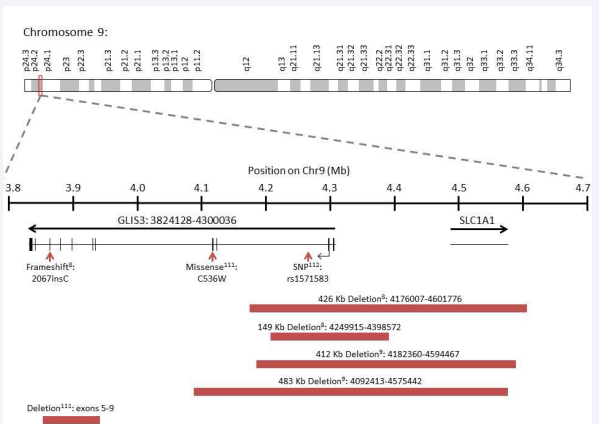
Figure 1: Summary of GLIS3 mutations associated with hypothyroidism in humans. Mutations in GLIS3 that are associated with hypothyroidism included a frame shift mutation resulting in premature termination (2067insC; 625FS703STOP), a missense mutation (C536W) in the zinc finger motif predicted to affect DNA binding, and several deletion mutations encompassing the 5’ UTR, exons 1-2, 1-4, or 5-9. A SNP in the second intron (rs1571583) has been associated with increased TSH and decreased T4 levels.Superscripts contains the reference numbers of the studies that described the indicated GLIS3 mutation.
Aside from the consistent presentation of neonatal diabetes and CH, additional features may include glaucoma, liver fibrosis, cystic kidneys, mental retardation, osteopenia, deafness, and pancreatic exocrine insufficiency. The variation in the GLIS3 phenotype is attributed to the tissue expression of variable length GLIS3 transcripts. Larger transcripts (7.5 kb) are predominately expressed in the pancreas, thyroid, and kidney, with smaller transcripts (0.8-2 kb) expressed in the heart, kidney, liver, and skeletal muscle; therefore, the presence of neonatal diabetes and CH is likely due to absence of the 7.5 kb GLIS3 transcript [8].
All NDH patients had high TSH and low T4 during the neonatal period; however, the absence of consistent pathological features makes it difficult to determine a causative mechanism. In most cases, patients have not responded to conventional treatment and maintained elevated levels of TSH despite normalization of T4. However, abnormalities in thyroid anatomy and/or T4 uptake are not sufficient to explain this. Three patients described by Senee et al. responded to daily T4 treatment, but subsequent TSH levels were not reported. Thyroid ultrasound and scintigraphy results also suggested athyrosis or hypoplasia with absent radioiodide uptake [8]. Patients described by Dimitri and Taha had high daily T4 requirements with persistently elevated TSH and increased TG levels despite normal thyroid anatomy. These patients don’t appear to be TSH resistant given that TSH values were reduced within the normal range after initial T4 supplementation [9,10]. For example, in one case of GLIS3 mutation with a deletion of exons 1-2, hypothyroidism was identified on day four of life with TSH levels >150 mIU/l and T4 at 4.3 pmol/l. The patient was treated with 20 µg/kg of T4 daily with sufficient TSH suppression. However, at two months of age, TSH levels exceeded 150 mIU/l and remained high despite and increased dose of 75 µg/kg daily T4. TG levels were also markedly elevated. A reduction in TSH was observed when T4 treatment was divided into four daily doses. No deficiencies in T4 uptake were present, and thyroid gland anatomy appeared normal on ultrasound examination [9]. In a meta-analysis of thyroid-related traits, a GLIS3SNP was associated with elevated serum TSH and decreased T4 levels [112]. Given that the mutations associated with GLIS3 result in heterogeneous clinical presentations of hypothyroidism, further research is required to elucidate the role of GLIS3 in the gene expression networks for thyroid development and hormonogenesis that contribute to CH.
SUMMARY AND CONCLUSIONS
A number of genetic mutations have been identified that result in developmental defects of the thyroid gland or defective thyroid hormone synthesis; however, the genes defective in many congenital hypothyroidism patients have yet to be identified. While it is clear that mutations in the gene encoding GLIS3 are associated with the development of CH, further research is needed to decipher the molecular mechanisms underlying its pathophysiology. Elucidation of the biological functions of GLIS3 in the thyroid will be crucial to the discovery of therapeutic opportunities for the treatment of CH.
ACKNOWLEDGEMENT
This research was supported by the Intramural Research Program of the NIEHS, NIH (Z01-ES-100485).









































































































































