Identification for Immunogenic Epitopes Candidates of CD16A Mimetics
- 1. Key Laboratory of Tropical Translational Medicine of Ministry of Education, School of Basic Medicine and Life Sciences, Hainan Medical University, China
Keywords
• CD16A; Immunogenic epitopes;Structural analysis
Citation
Wang Z, Fan S, Huang Z, Long Y, Shao J (2025) Identification for Immunogenic Epitopes Candidates of CD16A Mimetics.J Immunol Clin Res 8(1): 1053.
INTRODUCTION
CD16 A is a transmembrane protein with a short C-ter cytoplasmic tail and possesses two extracellular Ig like domains. CD16A displays ADCC potent killing effect competing with tumors and virus. For obtaining highly specific targeting CD16A antibodies, designing CD16A antigen appears to be particularly necessary so that enhancing combination between CD16A and anti-CD16A antibody and effectively recognizing CD16A binding sites.
CD16 A has 5 asparagine (N)-linked glycan at N38,N45,N74,N162,and N169. The N-glycans are composed of 23% of the high-mannose structure, 22% of hybrid type structure, and the remaining (55%), being complex type N-glycans. However, only the N162 N-glycan seems to directly impact CD16a affinity toward IgG, conversely with its position in the IgG-binding site. Although the N45 N-glycan is not situated in the IgG-binding site, it also contributes to the binding to IgG by stabilizing the CD16a structure. The Fc N297-linked glycan has a critical role in its interaction with CD16a, but less is known about the role of CD16a glycosylation and how it could impact the affinity for IgG.
Protein-protein interactions influence protein function by structural transform [1]. CD16 A interacts with the lower hinge/upper CH2 of IgG. The FcN-glycan chain linked to the N297 of the CH2 also plays a critical role in this interaction. The amino acid and the glycan composition can greatly influence the affinity of CD16a for the Fc and consequently the potency of ADCC. Recent data support that the Fab could be implicated in the IgG/CD16a interaction In addition to, mutations in the Fab highlight the potential interaction of Fab-CD16a during IgG-CD16a binding. Therefore, we analyzed and predicted the property, transmembrane domain, hydrophilic and hydrophobic and secondary structure of CD16A. Moreover,we also predict binding sites between CD16A and anti CD16A Fab.
MATERIALS AND METHODS
CD16A sequences retrieval
Sequence as the most foundational information, being used for analyzing characters. Therefore, we initially looked for the sequence of CD16A on NCBI database [2]. We eventually attained 8 sequences, as different isoform. Obtained sequences would be used for subsequent researches or analysis. The sequences were extracted from the database in the FASTA format.
B-cell epitopes
The BEs were divided into linear epitopes and conformational epitopes. To predict and map conformational B cell epitopes, the online Server BepiPred-2.0 (https://services.healthtech.dtu.dk/ service.php?BepiPred-2.0) was used to predict the linear B-cell epitopes (LBEs), and conformational epitopes with Discotope 2.0. Both prediction algorithms are available on the IEDB B cell prediction tool page (http://tools.iedb. org/main/bcell/). After obtaining the predicted epitopes from three different servers, we selected the most similar domain of sequences for further research [3,4].
Surface Accessible Regions
An ideal epitope should be conserved [1,2]. In order to determine the surface accessibility of the epitope, Emini surface accessible prediction tool of the IEDB was used. Epitopes that were found to have surface accessibility were selected and analyzed for conservancy. For the prediction of the conservancy, sequence identity cut-off was set to 90% [5-8].
Flexibility and hydrophilicity of B-cell epitopes
Hydrophilicity and flexibility of a peptide are related to its antigenicity. Selected B-cell epitopes were checked for their antigenicity by VaxiJen v2.0 server with a threshold value of 0.4.Conserved epitopes were submitted to Karplus and Schulz (KS) flexibility online tool, and Parker hydrophilicity prediction tool for flexibility and hydrophilicity predictions respectively.
Docking simulation assay
ZDOCK online web server was used to predict the binding spatial structure of these two proteins and in virtue of pymol tool to visualize the complex and calculated the interaction sites with its strength [9,10]. ZDOCK online web server requires two input proteins, we chose CD16A PDB file 5D6D and CD16A Fab PDB file 7SEG downloaded in protein data bank (PDB web) [11,12]. We selected chain C of 5D6D as binding portion, while selected chain A, B, C and D. The PDB files downloaded from ZDOCK web has been put to use for the next visual analyze. Pymol tool was utilized to forecast the protein-protein interaction and calculated the hydrogen bond length [13,14].
RESULTS AND DISCUSSION
Sequences aligning and conservation sites analysis
MEGA7 software was used to align all sequences (Figure 1), as well as Gblocks online software was used to analyze the conservation site among all 8 sequences (Figure 2).
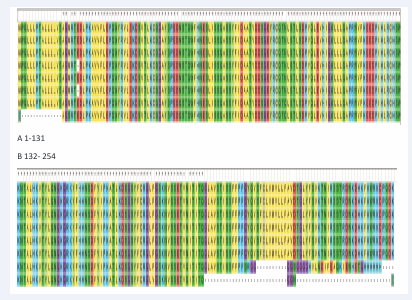
Figure 1: Sequences aligned by MEGA 7.10 arrays of sequences were aligned.(A)Amino acid position from 1-131;(B) amino acid position from 132-254.
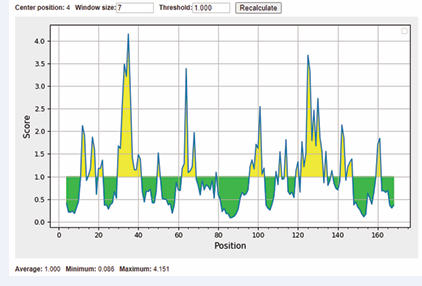
Figure 2: Surface accessible of sequences were showed. The x-axis and y-axis represented the sequence position and surface probability, respectively. The threshold value is 1.000. The regions above the threshold were antigenic, shown in yellow.
Utilization of MEGA 7 made sequences orderly arranged. Conservative sites were provided by Gblocks and underlined in blue line. As a result, the conservation sequences were selected and exhibited in Table 1. After aligning 8 arrays of sequences, we found that the redundancy causes the difference exists in three-type chains. Wiping of redundancy sequence and renumbering amino acid sequence, disappearing of E21 gives rise to distinction between the 254-amino acid-chain and the 253-amino acid-chain, which is exhibited in Figure 1.We subsequently predicted the conservation sites using Gblocks, except the second sequence mentioned above, any other sequences possess the same conservation sites which is tabulated later. We finally got 3 conservation sequences,C1,C2 and C3 [15].
Table 1: Conservative sequences predicted by Gblock
|
Sequence number |
Position |
Sequences |
|
Conservation 1 (C1) |
1-20 |
MWQLLLPTALLLLVSAGMRT |
|
Conservation 2 (C2) |
22-192 |
DLPKAVVFLEPQWYRVLEKDSVTLKCQGAYSPEDNSTQWFHNESLISSQASSYFIDAAT VDDSGEYRCQTNLSTLSDPVQLEVHIGWLLLQAPRWVFKEEDPIHLRCHSWKNTALHKVTYL QNGKGRKYFHHNSDFYIPKATLKDSGSYFCRGLFGSKNVSSETVNITITQ |
B-cell epitopes
We focused on potential B-cell epitopes for vaccine candidates. For the prediction of potential linear B-cell epitopes, three different software packages namely BepiPred 2.0 IEBD, BepiPred-2.0: Sequential B-Cell Epitope Predictor and ABCpred were utilized. We found that there were 10 common B-cell antigenic epitopes of proteins predicted by all the three prediction tools. The locations of these epitopes were 11–19,31–34,44–53,61–63,73–76,88–100,110–115,125–136,139–146 and 156–162 (Table 1). These identified epitopes were further comprehensively studied. Multiple studies have proved that an ideal epitope should be accessible to an antibody or a cell surface receptor (Tables 2-5) [16].
Table 2: Results of BepiPred-2.0 Sequential B-Cell Epitope Predictor
|
Sequence number |
NO. |
Start |
End |
Peptide |
Length |
|
C2 |
1 |
11 |
19 |
PQWYRVLEK |
9 |
|
2 |
30 |
34 |
YSPED |
5 |
|
|
3 |
45 |
53 |
LISSQASSY |
9 |
|
|
4 |
88 |
100 |
LLLQAPRWVFKEE |
13 |
|
|
5 |
110 |
115 |
WKNTAL |
6 |
|
|
6 |
125 |
136 |
KGRKYFHHNSDF |
12 |
|
|
7 |
139 |
146 |
PKATLKDS |
8 |
|
|
8 |
156 |
162 |
GSKNVSS |
7 |
Table 3: Results of ABCpred
|
Sequence number |
Rank |
Start |
End |
Peptide |
Length |
Score |
|
C2 |
1 |
51 |
64 |
SSYFIDAATVDDSG |
14 |
0.93 |
|
2 |
122 |
135 |
QNGKGRKYFHHNSD |
14 |
0.87 |
|
|
3 |
88 |
101 |
LLLQAPRWVFKEED |
14 |
0.85 |
|
|
3 |
17 |
30 |
LEKDSVTLKCQGAY |
14 |
0.83 |
|
|
4 |
80 |
93 |
QLEVHIGWLLLQAP |
14 |
0.81 |
|
|
6 |
29 |
42 |
AYSPEDNSTQWFHN |
14 |
0.78 |
Table 4: Common B-cell epitopes
|
Sequence number |
Start |
End |
Peptide |
Length |
|
C2 |
88 |
100 |
LLLQAPRWVFKEE |
13 |
|
125 |
135 |
KGRKYFHHNSD |
11 |
|
|
30 |
34 |
YSPED |
5 |
Table 5: Consensus sequences
|
Sequence number |
No. |
Sequence |
Length |
Start |
End |
|
C2 |
1 |
YSPEDNSTQWFH |
12 |
30 |
41 |
|
2 |
DSGEYRC |
7 |
62 |
68 |
|
|
3 |
VFKEEDPI |
8 |
96 |
103 |
|
|
4 |
QNGKGRKYFHH |
11 |
122 |
132 |
Surface accessibility
Then, we predicted surface accessibility using Emini Surface Accessibility Prediction tool. Our results showed that 2 of predicted surface accessible peptides possessed consensus sequences with the selected B-cell epitopes. These two surface accessible peptides were MRTEDL and QWYRVLEK. It is a pity that the common sequences of C2 and C3 between their surface accessible peptides were not found. As a result, these two peptides were used for the further research (Figure 3).
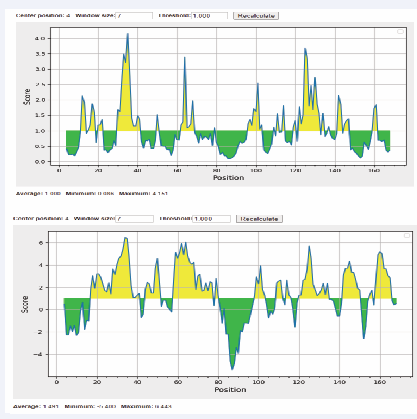
Figure 3: Surface accessible of sequences were showed.
Hydrophilicity
The online software AllerTOP 2.0 defined the CD16A as non-allergen. The VaxiJen 2.0 showed that the overall prediction value for the protective antigen was 0.6858,the virus was selected as the prediction model and the threshold for this model was 0.4.Therefore,CD16A was identified as safe protective antigen using allergenicity and antigenicity analysis [17].
Docking
We calculated the strength of hydrogen bonding of complex 1 and complex 2.There were 9 sites that 2 proteins possessed interaction force in complex 1.We listed the interaction sites between both proteins and the strength of hydrogen bonding between them in Figure 4 and table 6.
![CD16A-CD16A Fab complex. Nine pairs of interaction amino acids and CD16A Fab is S151-Y54, S164-Y98, T167-S31, E166-S31, E166-Y98, I88-Y98, I88-Y32, W90-A97, W90-S96 respectively. (A1-I1) overall situation of amino acids interaction in whole complex; (A2-I2) nine groups of interaction sites between 2 proteins detail situation. (light blue for CD16A, limon for CD16A Fab) [19].](https://www.jscimedcentral.com/public/assets/images/uploads/image-1744807613-1.PNG)
Figure 4: CD16A-CD16A Fab complex. Nine pairs of interaction amino acids and CD16A Fab is S151-Y54, S164-Y98, T167-S31, E166-S31, E166-Y98, I88-Y98, I88-Y32, W90-A97, W90-S96 respectively. (A1-I1) overall situation of amino acids interaction in whole complex; (A2-I2) nine groups of interaction sites between 2 proteins detail situation. (light blue for CD16A, limon for CD16A Fab) [19].
We found that I88, W90 and E166 could interact with multiple other amino acids, which indicated that these sites of amino acids could be the binding sites that interact with antibody. The structure revealed an extensive network of van der Waals interactions, ion pairs, hydrogen bonds, and additional elaborate coordination at the interface.The complex provided a structural rationale for the extreme sensitivity of the anti-CD16A Fab, and explained CD16A and its ability to bind the receptor in presence of competing IgG.Together, it could be the aim for designing structure domains of bispecific antibodies and CAR-NK cells [18].
Table 6: Interaction between CD16A and Fab
|
No. |
CD16A amino |
Anti-CD16A antibody amino |
Strength of hydrogen bond |
|
1 |
S151 |
Y54 |
2.5 |
|
2 |
S164 |
Y98 |
3.0 |
|
3 |
T167 |
S31 |
3.4 |
|
4 |
E166 |
S31 |
2.5 |
|
5 |
E166 |
Y98 |
2.7 |
|
6 |
I88 |
Y98 |
2.4 |
|
7 |
I88 |
Y32 |
2.9 |
|
8 |
W90 |
A97 |
2.5 |
|
9 |
W90 |
S96 |
2.9 |
DISCUSSION
Antigenicity, flexibility and surface accessibility are fundamental properties of an epitope which are essential to induce an immune response. We analyzed the flexibility and hydrophilicity of these antigenic epitopes, providing a critical theoretical foundation for targeted antibody design. Using MEGA7 and Gblocks tools, this study successfully identified three conserved sequences (C1-C3) of CD16A. Although these sites were not directly located within the IgG-binding region, its potential impact on the structural stability of CD16A required further validation through subsequent mutagenesis experiments. At threshold cutoff 1.0, the surface accessibility of CD16A was determined by Emini surface accessibility prediction tool. Our results showed two surface accessible peptides were MRTEDL and QWYRVLEK (Figure 2).
Identification of neutralizing B-cell epitopes will contribute to the development of a more effective universal vaccine to combat all diverse strains of HIV. With the aim to identify B-cell epitopes from HIV-1 diverse strains, immunoinformatics approaches allow us to primarily screen candidates of B-cell epitopes from a large number of sequences retrieved from the database. The conserved epitopes provided broader protection across multiple strains than epitopes derived from highly variable genomic regions. An ideal epitope should be highly similar or conserved. The conservancies of predicted B-cell epitopes were evaluated by the IEDB conservancy analysis tool. Our results showed that 2B-cell peptides showed 100% identity among all the strains sequences, indicating their cross-strain conservation. Such conserved epitopes could circumvent immune escape caused by high-frequency pathogen mutations, offering potential targets for broad-spectrum vaccine development. Next, these 2 highly conserved B-cell surface accessible peptides were scrutinized individually for the prediction of antigenicity (Table 3). We found that 3 common B-cell surface accessible peptides were highly antigenic (Table 4). Our work was in agreement with the previous study showing that prediction using two or more tools increased both accuracy and probability in defining a genuine B-cell epitope [19].
Evidence suggests that apart from antigenicity, flexibility and accessibility are two other fundamental properties of an epitope which are essential to induce an immune response.
Subsequently, we analyzed the flexibility and hydrophilicity of these antigenic epitopes. Flexibility was predicted using VaxiJen 2.0 prediction. These studies hence concluded that 3 linear B-cell epitopes were highly flexible and hydrophilic in nature. The overall prediction value for the protective antigen was 0.6858, and the threshold for this model was 0.4. Therefore, CD16A was identified as safe protective antigen using allergenicity and antigenicity analysis.
Protein docking simulations revealed key interaction sites between CD16A and Fab. Nine pairs of interaction amino acids and CD16A---Fab were S151-Y54, S164-Y98,T167-S31,E166-S31,E166-Y98,I88-Y98, I88-Y32,W90-A97,W90-S96 respectively. Moreover, I88 formed hydrogen bonds with Fab’s Y98/Y32,W90 interacted with A97/S96, and E166 bound to S31/Y98, suggesting these sites likely played pivotal roles in antibody binding. This study provided a structural foundation for bispecific antibodies and viral entry inhibitors against the HIV-1 envelope protein.
Taken together, Even though these B-cell epitopes are derived from a consensus sequence, some are highly conserved among the global PEDV strains, which represent a promising vaccine target for the development of a universal epitope-based vaccine as well as antibodies.
ACKNOWLEDGEMENTS
This study was supported by research grants, Hainan Provincial Key Research and Development Program (Grant No: ZDYF2022SHFZ077 and ZDYF2016126).
REFERENCES
-
HH, Chavez JD, Tang X, Bruce JE. Quantitative interactome analysis with chemical cross-linking and mass spectrometry. Curr Opin Chem Biol. 2022; 66: 102076.
-
Benson DA, Karsch-Mizrachi I, Lipman DJ, James O, Eric WS. GenBank. Nucleic Acids Res. 2009; 37: D26-D31.
- Grifoni Alba, John S, Yun Z, Richard HS, Bjoern P, Alessandro S, “A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2.” Cell Host Microbe. 2020; 27: 671-680.
- Mingkai Yu, Zhu Y, Li Y, Chen Z, Li Z, Wang J. “Design of a Recombinant Multivalent Epitope Vaccine Based on SARS-CoV-2 and Its Variants in Immunoinformatics Approaches.” Front immunol. 2022; 13: 884433.
- Singh J, Malik D, Raina A. Immuno-informatics approach for B-cell and T-cell epitope based peptide vaccine design against novel COVID-19 virus. Vaccine. 2021; 397: 1087-1095.
- Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985; 55: 836-839.Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, et al. The immune epitope database 2.0. Nucl Acids Res. 2010; 38: D854-D862.
- Bui HH, Sidney J, Li W, Fusseder N, Alessandro S. Development of an epitope conservancy analysis tool to facilitate the design of epitope- based diagnostics and vaccines. BMC Bioinformatics. 2007; 8: 361.
- Molini, Barbara, Mark CF, Godornes C, Vorobieva A, Lukehart AS, Giacani L. “B-Cell Epitope Mapping of TprC and TprD Variants of Treponema pallidum Subspecies Informs Vaccine Development for Human Treponematoses.” Front Immunol. 2022; 13: 862491.
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007; 8: 4.
- Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014; 30: 1771-1773.
- Seeliger DandDe Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput aided Mol Des. 2010; 24: 417-422.
- Ahmed AA, Keremane SR, Vielmetter J, Bjorkman PJ. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcγRIIIa. J Struct Biol. 2016; 194: 78-89.
- Kakiuchi-Kiyota S, Ross T, Wallweber HA, Kiefer JR, Schutten MM, Adedeji AO. A BCMA/CD16A bispecific innate cell engager for the treatment of multiple myeloma. Leukemia. 2022; 36: 1006-1014.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000; 28: 235-242.
- Kakiuchi-Kiyota, Satoko Kiefer JR, Schutten MM, Adedeji AO, et al. “A BCMA/CD16A bispecific innate cell engager for the treatment of multiple myeloma.” Leukemia. 2022; 36: 1006-1014.
- Ellwanger Kristina, Reusch U, Fucek I, Wingert S, Ross T. “Redirected optimized cell killing (ROCK®): A highly versatile multispecific fit- for-purpose antibody platform for engaging innate immunity.” MAbs. 2019; 11: 899-918.
- Molini, Barbara, Mark CF, Godornes C, Vorobieva A, Lukehart AS, Giacani L. “B-Cell Epitope Mapping of TprC and TprD Variants of Treponema pallidum Subspecies Informs Vaccine Development for Human Treponematoses.” Front Immunol. 2022; 13: 862491.
- Mingkai Yu, Zhu Y, Li Y, Chen Z, Li Z, Wang J. “Design of a Recombinant Multivalent Epitope Vaccine Based on SARS-CoV-2 and Its Variants in Immunoinformatics Approaches.” Front Immunol. 2022; 13: 884433.









































































































































{kind=link}