Effects of High-Salt Diet during Pregnancy on Fetal Nephrogenesis and Hypertension Later in Life: Aberrant Renin-Angiotensin System Programming
- 1. Department of Physiology, Wayne State University School of Medicine, USA
- 2. Department of Hypertension and Vascular Research, Ford Hospital and Wayne State School of Medicine, USA
- 3. Departments of Internal Medicine and Physiology, Wayne State University School of Medicine, USA
Abstract
It is widely accepted that excess salt intake is a risk factor for development of hypertension [1,2]. Current research also supports the idea that adverse conditions during pregnancy can affect progeny postnatal health, both immediately after birth and for the remainder of their lives [3,4]. The purpose of this mini review is to discuss the effects of a gestational high-salt diet on renal development and programming of the renin-angiotensin system in the fetus and how these changes increase the offspring’s incidence of hypertension and renal disease later in life.
After reviewing a large body of evidence collected from and related to maternal high-salt diet experiments, it is proposed that high salt (HS) diet during pregnancy causes an increased prevalence of hypertension in offspring due to {A} an overall increase in blood pressure resulting from absence of appropriate modulation of plasma renin activity in response to changes in salt concentration, {B} an increased pressor response to angiotensin II, and {C} a decrease in the number of glomeruli per kidney. (Figure 1) provides a summary of the pathophysiological changes in the fetus caused by maternal high HS diet, and how these changes lead to an increase in the rate of disease risk in offspring.
Citation
Provagna AJ, Beierwaltes WH, Rossi NF (2016) Effects of High-Salt Diet during Pregnancy on Fetal Nephrogenesis and Hypertension Later in Life: Aberrant Renin-Angiotensin System Programming. Ann Clin Exp Hypertension 4(1): 1032.
Keywords
• Blood pressure
• Renin
• Angiotensin
• Fetal programming
• Nephrogenesis
• Dietary sodium intake
ABBREVIATIONS
ACE: Angiotensin Converting Enzyme; Ang II: Angiotensin II; AT1 R: Antgiotensin Receptor Type 1; AT2 R: Angiotensin Receptor Type 2; AVP: Arginine Vaso Pressin; BUN: Blood Urea Nitrogen; CR: CReatinine; CAMP: Cyclic Adenosine Mono Phosphate; CGMP: Cyclic Guanosine Mono Phosphate; GFR: Glomerular Filtration Rate; HS: High Salt; JG: Juxta Glomerular; LS: Low Salt; NKCC: Sodium Potassium Chloride Co-transporter; NOS: Nitric Oxide Synthase; PRA: Plasma Renin Activity; RAS: Renin-Angiotensin System
INTRODUCTION
Health is dictated by genetics, the environment, and our behavior. The basic idea of fetal programming combines these factors; maternal behavior during pregnancy shapes the intrauterine environment and influences the health of offspring throughout their life. Fetal developmental programming is essentially the idea that maternal behaviors and traits like diet, exercise, hormone levels, stress, and drugs have an effect on the way a fetus develops. Alterations in fetus development, for example growth rate or specific protein levels, can then in turn cause the offspring to be more or less susceptible to certain diseases.
Fetal programming can be witnessed with a variety of environmental and nutritional variables encountered during development that can profoundly exert influences on physiological and pathophysiological functions in later life of the offspring. Among these are low protein intake, increased fat, dehydration, and high salt (HS) intake in the mother [4]. An increasing body of data suggests that these factors result in changes in gene expression and epigenetic modifications that may increase the risk of disease in offspring. A more complete understanding of these models of programmed hypertension could expose areas for potential intervention and possible drug targets, allowing clinicians to better control blood pressure in their patients. Improved control of blood pressure would decrease the incidence of renal failure, coronary heart disease, and other cardiovascular issues including stroke in the population. This could greatly impact the health of the population, as heart disease and renal failure are the first and eighth leading causes of death in the United States, respectively [5].
The Renin-Angiotensin System
In order to understand how a HS diet in utero can alter elements of the renin-angiotensin system (RAS) and lead to disease later in life, it is important to have a comprehensive understanding of RAS components and how its classical function is accomplished in vivo. The RAS is the primary, long-term regulator of systemic blood pressure homeostasis and, therefore, of renal perfusion [6]. In theory, low blood pressure causes decreased kidney perfusion thereby decreasing the salt load delivered to the macula densa. This results in renin release and an increase in blood pressure via elevated angiotensin II (Ang II), aldosterone, and arginine vasopressin (AVP) levels.
Angiotensinogen is the substrate for renin. It normally circulates in excess abundance, waiting to be cleared by the protease activity of renin. Angiotensinogen expression is upregulated by Ang II and sodium depletion. Its expression is also affected both negatively and positively by a variety of hormones [7].
The juxtaglomerular apparatus located near the glomeruli in the kidney acts as the salt sensor that controls renin release. The macula densa consists of specialized epithelial cells of the distal tubule. The juxtaglomerular (JG) cells are specialized cells primarily in the wall of the afferent arteriole that produce, store, and secrete renin. The macula densa and JG cells are in such close contact that their basement membranes are fused, allowing for quick, direct communication between the two cell types [8]. A decrease in blood pressure results in a decrease of renal perfusion and a decrease in the salt load delivered to the distal tubule that is sensed by the macula densa cells which use a variety of second messengers to cause an increase in cAMP in the JG cells. These high levels of cAMP cause renin release into the afferent tubule as cAMP is responsible for renin translation, renin mRNA stability, and renin exocytosis [9].
A decrease in salt load to the distal tubule is not the only signal that allows for release of renin. An increase in perfusion pressure will cause the afferent arteriole to stretch. This will in turn inhibit renin secretion. The sympathetic nervous system regulates renin release as well. Renal α adrenergic activity inhibits renin secretion while β adrenergic stimulation promotes renin release.
Once renin is released into the circulation it can cleave angiotensinogen into angiotensin I. Angiotensin I is inactive and must be further cleaved into Ang II by angiotensin converting enzyme (ACE). ACE is located on the apical membrane of endothelial cells throughout the body, but is most densely expressed in the lungs. ACE is a metalloprotease that utilizes zinc in order to perform its enzymatic activity [7]. ACE2 is a similar enzyme that is responsible for the break down Ang II.
Ang II utilizes two receptor types; the AT1 and AT2 receptors. The AT1 receptor can also be broken down into two subtypes: the AT1a and AT1b receptors. AT1a receptors are primarily vascularly localized while AT1b receptors are localized to the adrenal gland. AT2 receptors mediate many Ang II effects in the central nervous system and are expressed in high levels in the developing fetus. In the adult kidney, AT2 receptors are localized to large preglomerular arteries, while AT1 receptors are seen in arteries of all sizes, the glomeruli, and vasa recta [7].
Ang II exerts its classical functions via the AT1 group of receptors. It increases sodium reabsorption in the proximal tubule, thick ascending limb, and the beginning of the collecting duct by increasing the activity of the sodium-hydrogen exchanger. AT1 R activation in vascular smooth muscle causes vasoconstriction systemically. In regards to renal vasculature, the efferent arteriole is more sensitive to Ang II than is the afferent arteriole. Disproportionate vasoconstriction between the two arterioles helps to increase the filtered fraction of the blood that travels through each glomerulus. Ang II stimulates aldosterone release from the adrenal gland. Aldosterone, in turn, binds to mineralcorticoid receptors in the kidney, activates the epithelial sodium channel, and results in decreased sodium excretion. In addition, Ang II stimulates the hypothalamus to create a thirst response and increase AVP. AVP, also commonly referred to as anti-diuretic hormone, promotes water retention by activating the V2 receptors in the collecting duct and increasing water reabsorption via aquaporin 2. Thus, Ang II increases both sodium and water retention.
The Renin-Angiotensin System during Development
The human kidney begins to develop during the fifth week of pregnancy and continues to form up until the fortieth and final week of gestation [10]. Nephrogenesis begins with the ureter arising from the ureteral bud which induces mesenchymal cells to condense around the ureteral tips and differentiate into many different cell types [10,11]. The components RAS are expressed very early during nephrogenesis and at high levels. Renin is expressed in the human kidney by the 5th week of gestation, when nephrogenesis begins. The rat expresses higher levels of renin as a fetus than it does at any other point in its lifespan [7]. These high levels of RAS expression are not affected by maternal renin, as circulating renin does not cross the placenta [12]. The increased expression of renin is compounded by the fact that its half-life appears to be increased early in fetal development [7].
During early development, the RAS system performs different functions than it does in an adult. Without a fully developed vascular system, the endocrine functions of RAS are not as effective. Paracrine, growth modulating effects of RAS are the primary function at this time, with the endocrine functions developing concurrently with kidney growth. In the case of endocrine RAS functions, blood pressure control and ion homeostasis control appear first, and renal hemodynamic control including tubuloglomerular feedback, develop later [7]. RAS is necessary for the cardiovascular system develop in the fetus. Ang II has been shown to regulate development of cardiac myocytes and vasculature primarily via the AT2 receptor [4].
Expression levels and localization of RAS components are also very different in the developing fetus. Renin release in early development is coupled with branching of the vascular tree. Renin expression begins in the larger preglomerular arteries and slowly shifts to the afferent arterioles. Many experiments have been dedicated to understanding how this shift in renin expression is controlled [13]. Showed that the renin shift could not be prevented with loss of Ang II expression via ACE deletion. This demonstrated that the developmental renin expression shift is not under the control of Ang II. It appears more likely that microRNAs control the renin expressing cell phenotype [9] deleted Dicer, the microRNA processing enzyme, in renin-expressing cells. This deletion caused the loss of renin expression. Alteration of the microRNAs expressed along the renal vasculature could lead to the developmental renin shift and the ability for the arteriolar smooth muscle cells to revert to their previous renin-expressing phenotype. Regardless of how renin expression is controlled, it is clear that it is vital to the development of the renal vasculature, since ablation of reninsynthesizing cells during development results in small kidneys with atrophic glomeruli [14].
The expression levels of the two Ang II receptor subtypes are altered in the fetal kidney as well. While AT2 receptor (AT2 R) expression is low in the adult in comparison with AT1 R expression, 90% of the Ang II receptors expressed in the fetus are AT2 [7]. AT1 Rs are localized to the more mature glomeruli present deeper in the cortex, and AT2 Rs are in “the condensed mesenchyme and transiently in the newly formed nephrons” [10]. AT1 Rs promote extracellular matrix production, angiogenesis, cell differentiation, cell growth (hypertrophy) and cell replication (hyperplasia). AT2 Rs control apoptosis, mesenchymal to epithelial transformation, and growth control [11]. The distinct receptor functions of AT1 R and AT2 R are demonstrated by administration of low doses of losartan (AT1 R antagonist) and PD123319 (AT2 R antagonist) to pregnant rats late in gestation. “Losartan administration results in a disorganized nephrogenic zone, with immature glomeruli present throughout the kidney (versus restricted to the nephrogenic zone in control rats)” [11]. PD123319 administration causes a thickening of the nephrogenic zone and drastically increases the number of immature glomeruli present there [11]. The loss of organization of kidney structure with blockade of AT1 R illustrates its importance in cell differentiation and the regulation of tubular structure. The increase in size of the nephrogenic zone with the blockade of the AT2 R demonstrates its importance in the apoptosis of cells during nephrogenesis. These experimental results lead one to conclude that the AT2 R is important for early renal development, and the AT1 R becomes important later on in nephrogenesis. This correlates with the high expression of the AT2 R early in development and the eventual decline of AT2 R and increase of AT1 R expression during gestation.
Experimental alterations of RAS in utero, either via inhibitors or by mutations in genes, causes aberrant nephrogenesis “across a wide phylogenetic spectrum” [14]. Experimental inhibition of Ang II or its receptors results in severe renal structural irregularities [15]. For example, loss of the RAS during development has been shown to “result in severe vascular aberrations and kidney papillary hypoplasia” [16,9] described decreased numbers of glomeruli in response to Ang II inhibition during murine ontogeny. Vehaskari and Woods [4] administered losartan to inhibit RAS activity during a critical period in rat nephrogenesis and obtained similar results. The exposed progeny displayed hypertension. Clinically, loss of RAS components has been shown to have even more detrimental effects. The autosomal recessive condition renal tubular dysgenesis has been linked with mutations in any or all of the RAS components. Renal tubular dysgenesis causes persistent fetal anuria (inability to produce urine) resulting in oligohydramnios, a deficiency of amniotic fluid. Oligohydramnios causes many teratogenic effects including cranial anomalies, clubbed feet, pulmonary hypoplasia, and congenital joint contrastructures; referred to collectively as the Potter sequence [12]. Most affected progeny do not survive in utero, and those that do usually die soon after birth. Because loss of RAS components causes these and other developmental defects, it is important that expectant mothers take care not to partake in behaviors that may alter RAS expression in the fetus. For example, RAS inhibitors are often prescribed to lower blood pressure. However, all RAS inhibitors currently used pharmaceutically can cross the placenta and therefore should not be taken while pregnant or while attempting to conceive [14].
Experimental Models
Animal models of salt loading and hypertension are the principal sources for studies in this area. Rat and ovine (sheep) models are the two most commonly utilized for maternal HS experiments. In order to have an effect on nephrogenesis, salt loading must be administered at different times in gestation depending on the model used. Sheep gestation averages 147 days [17], with nephrogenesis occurring in the first trimester [4], starting at roughly gestational date 27 [18]. Rat gestation is much shorter, averaging 22 days [19]. Kidney development occurs during the second half of pregnancy in rats [4] and continues after birth until PD21 [20]. Because of this postnatal renal development, HS diets are continued throughout lactation until weaning at week 4 in the majority rat models.
It is advantageous that the two primary animal models in maternal HS and fetal programming experiments have “very disparate gestational lengths, kidney maturity at birth, and adult characteristics, as it strengthen the argument that the programming of hypertension is not species specific” [4].
Experimental Results
Experimental results are in reference to the offspring of dams fed various high, intermediate, and low salt diets while pregnant. For instance when referring to a “HS rat,” the rat in question is not a direct receiver of a HS diet, however it’s mother received a HS diet while pregnant with said offspring and throughout lactation after the offspring was born.
Variance among Findings: While a plethora of maternal HS diet experiments have been conducted, there is wide variance in results. The body of evidence on the results of maternal HS diet contains conflicting data. Increased blood pressure of offspring appears to be one of the only consistently repeatable experimental result in maternal HS diet protocols [21]. The cause of the conflicting results could be due to use of different animal models (sheep or rat), the duration and timing of salt exposure, the degree of the salt load [22], the gender of the offspring, or the use of different, and perhaps less exact in some cases, experimental methods for determining the same variables [21]. The degree of salt load is a specifically important factor as it has a notable effect on the palatability of the food for the pregnant rat. While dams receiving a HS diet containing 3% NaCl may eat just as much as dams receiving a normal salt diet (most often between 0.25% and 1% NaCl), dams fed HS diets reaching 8% NaCl will often eat less. This probable variance in caloric intake can introduce an additional variable into the experiment that has the potential to skew results.
Due to the variance in data between experiments, this minireview will relay the results of different experiments, focusing on similar findings but noting places where experiments conflict. Next, the aforementioned results will be combined to form a model of the best supported effects of maternal HS diet on programming of offspring, how they collectively predispose exposed offspring to hypertension and renal disease later in life, and their likely mechanisms.
Increased Blood Pressure: An increase in blood pressure in gestational HS diet models is often not seen immediately upon birth, but develops shortly after. In a rat model performed by [1], mean arterial pressure was significantly elevated in male HS offspring (3%) from postnatal month 5 until the end of their observation period (month 9). This significant increase is in comparison with an intermediate-salt (IS) group (0.51%). The average deviation between the groups from month 5 to month 9 was 7.8 mmHg. In a rat model performed by [23], HS (water replaced with 0.15M NaCl solution) offspring presented with higher systolic blood pressure (SBP) from PD30 to PD60 in comparison to the normal salt control group. Because SBP was only tested at PD30 and PD60, we do not know how early the difference in SBP occurred between the two groups. The difference in mean SBP was around 10 mmHg for both PD30 and PD60. This deviation was corroborated by the rat model performed by [24], their HS rats (3%) exhibited a mean arterial pressure that was an average of 10 mmHg higher than the intermediate (1%) and the basal (0.1%) groups over a period of seven weeks. [25] (HS = 8%, Normal Salt = 1.3%) and [21] (HS = 4%, Normal Salt = 0.26%) reported significant increases in blood pressure of HS rat males after the 8th week and 9th week of age, respectively. Interestingly, while [25] utilized a tail cuff method and saw increased blood pressure in both genders; [21] assessed blood pressure via implantation of a radiotelemetric probe and only saw increased blood pressure in male progeny. In contrast, [2] reported no difference in blood pressure between their HS (8%) and low salt (LS) (0.15%) rat groups.
Persistent Hypernatremia: In a rat model performed by [21], plasma sodium concentration was significantly increased (deviation of ~13 mmol/L) in offspring of HS versus normal salt groups after weaning. This phenomenon of persistent hypernatremia without postnatal dietary salt stimulus was not supported by other experimental models. Other investigators [1,2,22] described normal plasma sodium levels for all their experimental groups in the absence of a dietary salt stimulus.
Dietary Alterations: Descriptions of changes in diet due to maternal HS diet widely varied. While some experiments reported increased food intake in HS rats [1,2,26] reported decreased consumption of both food and water in HS sheep offspring [24] described an increased drinking reaction in response to Ang II in the HS group cited an increased preference for salt in adult HS rats, while [21] claimed there was no change in salt preference in adult HS rats.
Alterations in Body Weight/ Kidney: Body Mass Ratio: Difference in body weight was another variable producing inconsistent results between experiments [2] and [25] both noted that their HS offspring had significantly reduced body weight in comparison to NS controls. In contrast, [22] described an increase in body weight of the HS group resulting in a decreased kidney: body mass ratio. Other investigators [1,16, 17,24] reported no significant difference in body weight between experimental groups.
Decreased Glomerular Number and Glomerulomegaly: [1] Reported a 39% reduction in the number of glomeruli present in each kidney in rat HS offspring (12,300 per kidney ± 2,500) versus IS offspring (31,600 ± 4,300). They also reported a 9-fold increase in average glomerular volume in HS offspring (6.91 ± 2.72 x 106 µm3 ) versus IS offspring (62.25 ± 0.89 x 106 µm3 ). [17] reported sheep HS offspring had 20% fewer glomeruli per kidney in comparison with control progeny. HS offspring had glomeruli with a radius 18% larger and a cross-sectional area 19% larger than control glomeruli. This phenomenon of a small number of large nephrons is referred to as oligomeganephronia [25] and Vehaskari and Woods [4] reported no change in the number of glomeruli between experimental groups. While the technique for estimating the glomerular number was not indicated by Vehaskari and Woods [4], both [25] and [1] utilized the fractionator method (the “gold standard” for estimating glomeruli number). The difference in results between these two experiments is not readily apparent, but may be due to use of different rat species or dietary salt concentrations.
Changes in Creatinine and Blood Urea Nitrogen: In the rat experiment carried out by [1], creatinine serum levels were decreased in HS offspring and creatinine clearance was increased compared with NS controls. In the ovine experiment carried out by [22], fetal blood urea nitrogen (BUN) was significantly increased in the HS group and BUN: CRE serum ratio increased in the HS group both during gestation and postnatally. A decrease in creatinine serum levels in the HS offspring are likely related to hyperfiltration and glomerulomegaly.
Changes in Protein Levels Related to Nephrogenesis: [1] Reported decreased levels of renin and AT1 R in HS offspring at term. However, in one week old offspring an increase in the protein levels of renin, ACE, and AT1 R, and a decrease in the level of AT2 R were observed in comparison with other groups [22] reported an increase in HS fetal angiotensinogen, ACE, AT1 R, and AT2 R protein levels with an increase in the ACE: ACE2 expression ratio in renal cortical tissue. They also saw an increase in the AT1 :AT2 receptor ratio in HS offspring postnatally in renal cortical tissue. [25] reported an increase in renal expression of Ang II in HS progeny. Overall, RAS-specific protein levels seem to be increased in offspring after exposure to a HS diet during pregnancy.
Investigators have looked at protein levels for a number of factors related to hypertension or nephrogenesis in offspring after gestational HS exposure [1] reported an increase in the protein levels of WT-1, sprouty1, and GDNF (glial cell line-derived neutrophic factor), and a decrease in FGF-2, PAX-2, and VEGF in one week old HS offspring. Changes in any of the aforementioned proteins can prove detrimental for nephrogenesis as they are all expressed at different stages in and are required for renal development. The decrease in FGF-2, VEGF, and PAX-2 is concerning as FGF-2 promotes nephron development and prevents mesenchyme tissue apoptosis and VEGF promotes angiogenesis. PAX-2 is necessary for the mesenchymal tissue to differentiate into endothelium [1,23] described that a decrease in Ang II as a result of gestational HS diet causes a reduction in the protein expression of α-SM-actin, proliferating cell nuclear antigen, and fibronectin in the renal cortex. Alpha-SM-actin is vital for cell structure and integrity. Proliferating cell nuclear antigen and fibronectin are important players in cell growth, replication, and differentiation. [2] reported that gestational HS intake was correlated with increased TGF-β production in glomerular endothelial cells. TGF-β is an anti-proliferative and pro-apoptotic chemokine. Thus, a number of factors involved in development appear to be influenced by exposure to a maternal HS diet.
Attenuated Modulation of Plasma Renin Activity: Attenuation of plasma renin activity (PRA) modulation by maternal HS diet was strongly supported experimentally. Under normal circumstances, if an individual is given a HS load, they should experience a decrease in PRA. [25] described that while low and normal salt offspring exhibited a decrease in PRA in response to salt loading, the HS group did not. [21] reported HS progeny, contrasted with control animals, had increased sodium excretion in low salt conditions and retention in HS conditions. This seemingly paradoxical and aberrant sodium handling suggests a similar lack of PRA modulation in response to sodium conditions [26] also looked in to modulation of PRA in response to a salt dose in pregnant ewes. After both 25 gram and 50 gram oral salt doses, the HS offspring decreased PRA only 50% as much as NS offspring.
Increased Angiotensin II Pressor Response: Both [27] and [24] cited an increased presser response to Ang II in HS offspring, meaning the same dose of Ang II caused a greater increase in MAP in HS versus NS rats. While Arugelles et al. did not provide quantitative results and instead only represented this increase in Ang II pressor response in bar graph; [24] described a 13.1 mmHg greater increase in pressor response in HS offspring in comparison to IS offspring. This response was measured in the carotid artery of rats in response to a 100µg/kg Ang II subcutaneous injection. While only two experimental protocols reported increased Ang II pressor response in HS offspring, they were the only two reviewed that tested for it. No experimental data reviewed showed evidence to the contrary. These results suggest enhance vascular reactivity to Ang II in offspring of HStreated mothers.
Proteinuria: Proteinuria, specifically albuminuria, was seen in HS groups in multiple experiments [1], saw significantly higher urine albumin secretion in male HS rats versus IS rats from month 3 until the end of observation. This albuminuria was greatly exacerbated by placing the HS offspring on a HS diet. These exact findings were corroborated by [2], with significantly higher albuminuria in HS rats, with an increase in urine albumin excretion seen with postnatal HS diet [17] reported conflicting findings in their sheep model, with both the HS and NS groups showing similar amounts of protein leakage into Bowman’s space, the tubule, and collecting duct lumina.
Increased Risk of Renal Injury and Phagocyte Infiltration: Renal histology suggests maternal HS diets can lead to damage of fetal kidneys. [23] reported that HS rodents had more glomerulosclerosis and tubulointerstitial lesions in the renal cortex when scored histological after insults such as a laparotomy and manipulation of the renal pedicles. This increase of incidence in renal injury was coupled with an increase in phagocyte infiltration.
DISCUSSION
Widely variable and conflicting data reported in changes in food intake, water intake, preference for sodium, and body weight make it difficult to dram any definitive conclusions regarding these areas when analyzing the body of evidence associated with high maternal salt intake during pregnancy. However, consistent results suggest a HS diet during pregnancy causes {A} an overall increase in blood pressure (hypertension) due to absence of PRA modulation in response to changes in salt concentration, {B} an increased pressor response to Ang II, and {C} a decrease in the number of glomeruli per kidney. Each of these is discussed in detail below {A} Absence in PRA modulation in response to changes in dietary salt concentration would maintain high blood pressure by increasing sodium retention during salt loading and decreasing sodium excretion during salt restriction. The shift to a higher blood pressure could be a result of increased renal expression of Ang II [25]. This is corroborated by documentation of increased Ang II expression in the kidneys of HS offspring [25]. These increased Ang II levels could be a result of the increase in the ACE: ACE2 expression ratio in the kidney [28]. This is because ACE works to form Ang II from angiotensin I; while, ACE2 breaks down Ang II. The increase of ACE and the decrease of ACE2 would favor high levels of Ang II. Research discovering and increase in the ACE: ACE2 ratio in fetal HS kidneys [22] and an increase in ACE levels in the kidneys of one week old HS offspring [1] support this theory. Therefore, an alteration in ACE and ACE2 levels could be responsible for an increase in Ang II expression, the shift to a higher blood pressure, and consequently the lack of normal PRA modulation seen in HS offspring.
The lack of PRA modulation could be due to alterations in RAS protein expression caused by changes in the epigenome. Alterations of maternal diet have been shown to affect the development of disease in offspring by modifying methylation and acetylation of chromatin structure [3]. Both ACE and AT1 R have CpG islands in their promoter regions [14]. CpG islands are areas of DNA were differential methylation takes place in order to turn a gene on or off. Increased levels of ACE and AT1 R expression in offspring exposed to a HS diet during gestation has been documented on more than one occasion [1,22]. It is possible that the maternal HS diet, caused demethylation of the CpG islands in the promoters of both of these genes, promoting their translation; however this has not yet been shown experimentally. In fact, a gestational low protein diet has been shown to cause a reduction in Ang II receptor gene methylation [14]. It is thus possible that other diet alterations during pregnancy, including a HS diet, may alter methylation of these genes as well.
{B} The increased pressor response to Ang II could result from increased AT1 R protein expression in the vasculature. Increased AT1 R expression was seen in the kidneys of several HS diet experimental progeny [1,22]. Experimental proof of localization of this increased expression to the luminal side of the endothelium of renal and systemic vasculature would be ideal. This increase in AT1 R protein expression explains the increased drinking in response to Ang II described in [24], as AT1 R is the receptor in the subfornical organ of the brain that controls the thirst response.
{C}A decrease in the number of glomeruli has been cited in humans with hypertension [1]. The decrease in glomerular number could be due to a number of causes. Increased TGF-β production in glomerular endothelial cells is consistent with renal hypoplasia, as TGF- β is anti-proliferative and can induce apoptosis. A reduction of fetal RAS activity due to fetal hypernatremia may also deny the developing kidney of important nephrogenic regulation, leading to hypoplasia [21] showed in vitro that increased extracellular salt impaired kidney growth, lending the idea that fetal hypernatremia could impair kidney growth in vivo. Although they did not see fetal hypernatremia in their in vivo model, both [29] and [22] were able to demonstrate that maternal HS intake during pregnancy can cause an increase in fetal plasma sodium concentration. This would support the hypothesis that maternal HS intake can cause aberrant nephrogenesis via a decrease in local RAS activity in response to increased plasma sodium levels in the fetus. The decrease in VEGF levels that has been documented in HS offspring [1] is also consistent with a decrease in glomerular number, as VEGF is pro-angiogenic and has its expression increased by Ang II. A reduction in fetal RAS would decrease Ang II, and then decrease VEGF, resulting in decreased angiogenesis and therefore fewer glomeruli.
Glomerulomegaly is likely a result of the decreased number of nephrons attempting to maintain GFR due to the resultant decrease in filtering surface area. With a decrease in the number of glomeruli, the remaining glomeruli are forced to filter more of a load. This increase in the filtration fraction of each glomerulus causes tubule damage and glomerular fibrosis, leading to an even further reduction in nephron number. This “hyperfiltration” of the glomeruli could explain the increased creatinine clearance seen in HS rats reported by [1]. The increased incidence of glomerulosclerosis seen by [23] is consistent with the association of decreased nephron number with development of glomerulosclerosis later in life [17].
Several experiments reported results with a gender effect [1], HS male rats had a more pronounced increase in MAP over female HS rats. [21] Found an even more interesting result, with HS males showing an increase in BP over NS rats and HS females showing a decrease in BP compared to the control. Other experimental models of programmed hypertension show similar discrepancy between genders, with hypertension proving more pronounced in males in food restriction models [4]. It is possible females have a protective mechanism, resulting in the need for increased environmental insult to produce the same effect seen in males. Some investigators have hypothesized estrogen is responsible for providing females with protection against hypertension [21], but this has yet to be demonstrated.
CONCLUSION
The purpose of this minireview was to discuss the effects of a gestational HS diet on renal development and programming of RAS in the fetus; and how these changes increase the offsprings’ incidence of hypertension and renal disease later in life. This discussion is vital due to the high prevalence of hypertension, kidney disease, and heart disease in the U.S. population and the high sodium content of processed food and fast food that is popular in the United States.
This review theorized a maternal HS diet during pregnancy causes aberrant RAS programming and therefore increased prevalence of hypertension and renal disease in offspring due to {A} an overall increase in blood pressure due to absence of PRA modulation in response to changes in salt concentration, {B} an increased pressor response to Ang II, and {C} a decrease in the number of glomeruli per kidney. These changes in offspring are supported by various maternal HS diet experimental protocols. The attenuated PRA modulation and increased pressor response to Ang II are results of abnormal RAS protein expression levels, including increased levels of ACE and AT1 R caused by decreased methylation in the CpG islands in the promoter regions of their genes. The decrease in the number of glomeruli per kidney in offspring exposed to a HS diet prenatally is likely a result of the loss of important nephrogenic regulation due to fetal hypernatremia, altered levels of RAS, and therefore levels of proteins that RAS regulates, including VEGF and TGF-β (Figure 1)
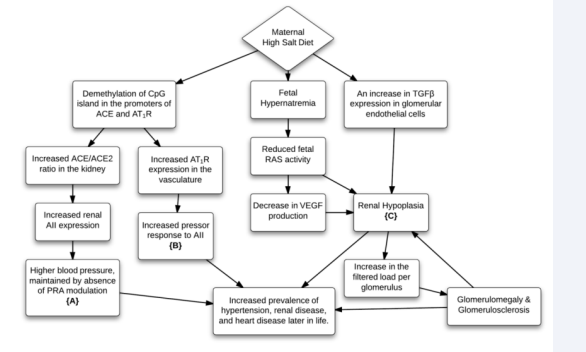
Figure 1: Effects of a Gestational High Salt Diet. The diamond represents maternal behavior, whereas the rectangles are representative of effects in the offspring. Beginning at the diamond, this figure depicts the proposed mechanisms for how gestational high salt (HS) diet results in increased prevalence of disease in offspring. Three rectangles include a notation of either {A}, {B}, or {C} differentiating them as the effects of a HS diet in utero that lead to the increased prevalence of hypertension in the offspring.
depicts the interplay of all the pathological changes seen in the fetus in response to a HS diet in utero and how these changes result in the increased incidence of hypertension and other life threatening diseases in offspring.
A wide variance of results was seen in maternal HS diet and programmed hypertension research. By comparing many experimental models dedicated to both the effects of aberrant RAS and of maternal HS diet on fetal renal development and increased incidence of hypertension later in life it has been possible to develop a strong hypothesis regarding the alterations resulting from maternal HS diet in utero and the likely mechanisms causing these changes.
Future Directions: Future research should be focused on determining the cause of the abnormal, blunted PRA modulation in response to changes in salt concentration and the consequences of the various protein variations seen in HS offspring. Specifically, many of the changes in RAS protein levels could be better evaluated with a more complete understanding of how each RAS component affects nephrogenesis and of the second messengers responsible for their growth modulation effects. Further research should also be focused on the methylation levels of the ACE and AT1 R promoters in response to a maternal HS diet as compared to the low protein diet research. It might also be of interest to elucidate the possible mechanisms whereby gonadal hormones may modulate these effects and protect against hypertension.
ACKNOWLEDGEMENTS
This review was funded in part by a grant by the Veterans Administration to NFR.
REFERENCES
5. Leading Causes for Death in the United States. Centers for Disease Control and Prevention. 2013.
8. Barajas L. Anatomy of the juxtaglomerular apparatus. Am J Physiol. 1979; 237: 333-343.
10. Gomez RA. Role of angiotensin in renal vascular development. Kidney Int Suppl. 1998; 67: 12-16.
17. Schoenian S. Sheep 201: A Beginner’s Guide to Raising Sheep. 2012.









































































































































{kind=link}