Survivorship Analysis for Osteosarcoma and Ewing
- 1. Pediatric Hematology-Oncology, Department of Pediatrics, LSUHSC/Children’s Hospital of New Orleans, USA
- 2. Department of Pathology, LSUHSC/Children’s Hospital of New Orleans,USA
- 3. Department of Radiology, Children’s Hospital of New Orleans, USA
- 4. Department of Biostatistics, LSUHSC New Orleans, USA
Abstract
Primary malignant bone tumors account for approximately 6% of all childhood malignancies. Osteosarcoma (OS) and Ewing’s sarcoma (ES) are the most common and have an annual incidence of 8.7 per million under the age of 20 years. The mean 5-year survival for OS and ES has been 61% and 60% respectively. The survival for OS and ES has not significantly improved for past 20 years.
We examined the cases of OS and ES, treated at Children’s Hospital of New Orleans (CHNOLA) from 1999-2012. Comparison to national survival data from the Survival, Epidemiological and End Results (SEER) study was done. The goal of our study was to demonstrate any difference in survival of our patient population compared to national data.
Results: Of the 44 patients diagnosed and treated as either OS or ES, 25 (57%) were OS and 19 (43%) were ES. The mean age of diagnoses for OS was 14 years and for ES was 12 years. Overall survival for all cases was 85%, which was superior to the reported SEER 5-year survival of 68.7 % for malignant bone tumors for ages 0-19 years from 2003 to 2009.
Survival was not affected by patients’ age, gender, race or timing of surgery for removal of primary tumor. All non-survivors had metastatic disease at presentation, which was an adverse prognostic factor (p=0.002). Additionally, positive tumor margins at time of surgery (p=0.008) and decreased amount of tumor necrosis post chemotherapy (p=0.001) negatively affected survival.
Conclusion: Overall survival of pediatric patients with bone tumors was better than SEER data. Presence of metastatic disease and poor response to chemotherapy based on tumor necrosis and positive margins were found to be adverse prognostic factors.
Keywords
• Bone tumors
• Osteosarcoma
• Ewing’s Sarcoma
• Survival
Citation
Raulji C, Pritchett H, Stark M, Ward K, Cruz V, et al. (2014) Survivorship Analysis for Osteosarcoma and Ewing’s Sarcoma in Children and Adolescents at a Single Pediatric Institution: Comparison to SEER Data. Ann Pediatr Child Health 2(1): 1011.
Introduction
Primary malignant bone tumors account for approximately 6% of all childhood malignancies. Of these, osteosarcoma and Ewing’s sarcoma are the most common and have an annual incidence of 8.7 per million under the age of 20 years [1]. Osteosarcomas and Ewing’s sarcomas comprise 56% and 34% of all malignant bone tumors, respectively [2].
Osteosarcomas are derived from primitive bone-forming mesenchymal stem cells and most often occur near the metaphyseal portions of long bones. Most osteosarcomas occur in the extremities, with the most common sites being the distal femur, the proximal tibia, and the proximal humerus [1]. About 80% of cases have localized tumor at presentation whereas the remainder present most commonly with pulmonary metastasis [1]. Treatment generally includes high dose methotrexate, doxorubicine and cisplatin, with some regimens adding ifosfamide and etoposide [1,3]. Neoadjuvant chemotherapy is given for 3 months prior to surgical resection of the tumor. Histologic response to preoperative chemotherapy has been identified as an important prognostic factor [3]. Long term disease-free survival for non metastatic osteosarcoma can be expected in the range of 60 to 75 % [4]. In contrast, it is only 10% to 30% for patients with metastases [4].
Ewing’s sarcoma is the second most common malignant bone tumor of childhood and adolescence. It is a small, round, blue-cell tumor thought to arise from neural crest cells [1]. It is evenly distributed between the axial and appendicular skeleton, and, in the long bones, the diaphysis is its most typical location [1,5]. Treatment includes an alternating regimen of chemotherapy drugs (vincristine-doxorubicin-cyclophosphamide and ifosfamide-etoposide) and interval compression for localized disease [6]. Non-metastatic disease at presentation has a 5-year disease-free survival rate of 70% whereas patients with metastases have a poor prognosis with a survival rate of approximately 25% [1,5].
Objectives
To examine all cases of osteosarcoma and Ewing’s sarcoma treated at Children’s Hospital of New Orleans (CHNOLA) between 1999-2012 and compare our survival and other characteristics to national data from the 2010 Survival, Epidemiological and End Results (SEER) study.
Study Design
A 13-year retrospective analysis of medical records for all patients diagnosed with osteosarcoma or Ewing’s sarcoma and treated at CHNOLA was performed. Basic information including patient age, sex, race, date of diagnosis and type of diagnosis was recorded. Data was also collected regarding the type of treatment received, surgery, response to treatment prior to surgery, date of relapse (if any) and survival. Statistical analysis was performed to determine correlation between clinical variables and overall/ disease-free survival and comparison was made to SEER data from 2010.
Results
From January 1, 1999 to December 30, 2012, 44 patients were diagnosed and treated for either Osteosarcoma or Ewing’s sarcoma at our institution. Twenty five of the cases were osteosarcoma and 19 were Ewing’s sarcoma. Demographic information from the cohort is presented in (Table 1).
Table 1: Patient Demographics.
|
Demographics |
Disease |
p value |
|||
|
Total N (%) |
OS N (%) |
ES N (%) |
|||
|
Age |
Mean (Years) |
12.75 |
13.4 |
11.9 |
0.275 |
|
SEX |
Male |
20 (45%) |
12 (48%) |
8 (42%) |
0.697 |
|
Female |
24 (55%) |
13 (52%) |
11 (58%) |
||
|
RACE |
African-American |
15 (34%) |
13 (52%) |
2 (11%) |
0.015 |
|
Caucasian |
26 (59%) |
11 (44%) |
15 (79%) |
||
|
Other |
3 (7%) |
1 (4%) |
2 (10%) |
||
|
METASTASIS |
No |
23 (53%) |
13 (52%) |
10 (56%) |
0.818 |
|
Yes |
20 (47%) |
12 (48%) |
8 (44%) |
||
|
RELAPSE |
No |
33 (84%) |
21 (88%) |
12 (77%) |
0.528 |
|
Yes |
6 (16%) |
3 (12%) |
3 (23%) |
||
|
Necrosis At Surgery |
<90 % |
7 (23%) |
6 (27%) |
1 (12%) |
0.398 |
|
>90 % |
23 (77%) |
16 (73%) |
7 (88%) |
||
|
Margins |
Negative |
28 (82%) |
17 (77%) |
11 (92%) |
0.201 |
|
Positive |
6 (18%) |
5 (23%) |
1 (8%) |
||
|
Overall Survival |
Alive |
33 (85%) |
20 (83%) |
13 (87%) |
0.779 |
|
Dead |
6 (15%) |
4 (17%) |
2 (13%) |
||
N: Number of patients; OS: Osteosarcoma, ES: Ewing’s Sarcoma.
Of note, Ewing’s sarcoma was significantly more prevalent in caucasian patients (p= 0.015), which is consistent with national norms. The numbers of patients with and without metastasis were evenly distributed in our patient cohort, and were not statistically significant.
Overall survival for all cases was 85%, with 83% for osteosarcoma and 87% for Ewing’s sarcoma. This was superior to the reported SEER 5-year survival of 68.7 % for malignant bone tumors for ages 0-19 years from 2003 to 2009. Figure 1
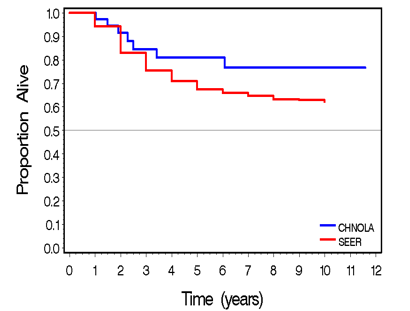
Figure 1 Kaplan-Meier curve comparing relative survival for all bone tumor patients at CHNOLA and SEER.
shows comparison of outcomes by Kaplan-Myers curves, between our cohort and the available SEER data.
Furthermore, survival was not affected by patients’ age, gender, race or timing of initial surgery. All non-survivors had metastatic disease at presentation, which was a statistically significant adverse prognostic factor (p<0.01). Additionally, positive margins after surgery, relapse and poor tumor necrosis post-chemotherapy negatively affected survival (Table 2).
Table 2: Factors Affecting Overall Survival in Bone Tumors.
| Demographic | N | % survival | p value | |
| Gender | Male | 18 | 94 | 0.189 |
| Female | 21 | 76 | ||
| RACE | White | 22 | 82 | 0.69 |
| African American | 14 | 86 | ||
| Other | 3 | 100 | ||
| Diagnoses | OS | 24 | 87 | 1 |
| ES | 15 | 83 | ||
| Time of Surgery | At Diagnoses | 4 | 100 | 0.296 |
| <3 months | 22 | 77 | ||
| >3 months | 9 | 100 | ||
| Metastasis | Yes | 17 | 65 | <0.001 |
| No | 21 | 100 | ||
| Relapse | Yes | 6 | 33 | <0.001 |
| No | 31 | 94 | ||
| Margins | Positive | 6 | 33 | <0.001 |
| Negative | 27 | 96 | ||
| Necrosis | > 90 % | 22 | 100 | <0.001 |
| < 90 % | 7 | 29 | ||
N: Number of Patients; OS: Osteosarcoma; ES: Ewing’s Sarcoma.
Discussion
Major advances have been achieved in the treatment of osteosarcoma with the discovery of several chemotherapeutic agents that are active in this disease as well as improvements in local control and limb-sparring surgical modalities [7]. For non-metastatic osteosarcoma, the efficacy of surgery in combination with systemic chemotherapy is well established [8]. Similarly, the treatment of Ewing’s sarcoma typically begins with neoadjuant chemotherapy. Local control is then addressed with surgery, radiation therapy, or a combination of the two modalities. Additional adjuvant chemotherapy is then used after local control.
A number of studies have shown better outcomes in survival when adolescents are treated at pediatric oncology centers compared to their adult counterparts [9-11]. All the patients in our study cohort were treating at a freestanding children’s hospital. Most patients were treated per Children’s Oncology Group protocols or by following strict pediatric oncology standards of care. This in turn may have contributed to our cohort having better survival rates than the national average.
Previous data has shown that having metastatic disease is one of the most important factors affecting survival in bone tumors [4,12,13]. Other relevant factors include poor histological response to chemotherapy, inadequate surgical margins and relapse [14,15]. We were able to duplicate these findings, again demonstrating the importance of local control for the treatment of these malignant tumors. All the patients in our cohort had surgery done by pediatric orthopedic surgeons who had expertise in these procedures, which in turn may have translated into good local control and possibly better survival when compared to SEER data.
Our study was limited by the fact that it was a retrospective review. We also acknowledge the fact that our patient cohort was far smaller than the SEER cohort. Additionally, some of the patients in our review were diagnosed recently and may not have enough follow-up time, and this could have contributed to the better survival seen at our institution.
In summary however, our data did show significantly improved outcomes for bone tumor patients when treated at a pediatric oncology center with a highly specialized comprehensive team that included orthopedic surgeons with expertise in advanced local control techniques. More effective therapies, perhaps targeting biological markers, are still needed for patients with metastatic disease.









































































































































{kind=link}