Arterial Mechanics, Extracellular Matrix, and Smooth Muscle Differentiation in Carotid Arteries Deficient for Rac1
- 1. Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, USA
Abstract
Stiffening of the extracellular matrix (ECM) occurs after vascular injury and contributes to the injury-associated proliferation of vascular smooth muscle cells (SMCs). ECM stiffness also activates Rac-GTP, and SMC Rac1 deletion strongly reduces the proliferative response to injury in vivo. While these resultss strongly implicate Rac in the stiffness-dependent SMC proliferation, ECM stiffening and Rac also affect SMC differentiation, which, in itself, can influence cell stiffness and proliferation. Here, we used immunofluorescence analysis and pressure myography of mouse carotid arteries to interrogate the effect of in vivo Rac1 deletion on SMC differentiation and arterial stiffness. The results show that medial abundance of alpha-smooth muscle actin or smooth muscle-myosin heavy chain, markers of the SMC differentiated phenotype, were not statistically different in carotid arteries containing or deficient in SMC Rac1. Nor did Rac1 deficiency have a statistically significant effect on carotid artery contraction to KCl, a functional readout of the contractile SMC phenotype. Similarly, the abundance of arterial collagen-I, -III, or -V, the integrity of arterial elastin, or arterial responses to pressure, including the axial and circumferential stretchstrain relationships that are assessments of arterial stiffness, were unaffected by deletion of SMC Rac1. Overall, these data argue that the inhibitory effect of Rac1 deletion on in vivo SMC proliferation reflects a primary effect of Rac1 signaling to the cell cycle and is independent of changes in SMC differentiation state or arterial stiffness.
Keywords
• Myography
• Arterial stiffness
• Carotid arteries
CITATION
Assoian RK, Xu T, Roberts E (2024) Arterial Mechanics, Extracellular Matrix, and Smooth Muscle Differentiation in Carotid Arteries Deficient for Rac1. Ann Vasc Med Res 11(1): 1178.
INTRODUCTION
Large elastic and muscular arteries such as the carotid and femoral contain a single-cell layer of endothelial cells (called the intima) that provides a non-thrombogenic surface around the lumen, a thick layer of vascular smooth muscle cells (SMCs) (called the “media”), and a diffuse outer layer (called the “adventitia”) that typically contains fibroblasts, inflammatory, and precursor cells [1-4].
Vascular SMCs are highly plastic and can undergo reversible differentiation/ de-differentiation [5-8]. In vivo, arterial SMCs normally exist in a differentiated (also called “contractile”) state characterized by periodic contraction, quiescence, and high expression of contractile proteins such as smooth muscle myosin heavy chain (SM-MHC) and alpha-smooth muscle actin (α-SMA). However, SMC de-differentiation is a hallmark of several vascular pathologies including atherosclerosis and the response to vascular injury. This de-differentiated (also called “synthetic”) phenotype is characterized by reduced contraction and an increased capacity to proliferate and migrate. Additionally, remodeling of the extracellular matrix (ECM) occurs during SMC de-differentiation to the synthetic phenotype and is thought to contribute to the arterial stiffening seen after vascular injury and in atherosclerosis [9,10]. Arterial stiffening, in turn, is permissive for cell proliferation and migration [9-11].
While the arterial ECM is highly complex, several studies have shown that two of the best studied components, elastin and the fibrillar collagens, have distinguishable roles in arterial mechanics. Elastin allows for arterial recoil and dominates arterial stiffness at low stress and stretch while fibrillar collagens (mostly collagen-I) regulate arterial stiffness at higher stress and stretch [2]. In addition to collagen-I, arteries also express lower amounts of collagen-III and collagen-V, but the exact roles of these fibrillar collagens in arterial stiffening is still incompletely understood. Nevertheless, altered expression, degradation and crosslinking of fibrillar collagens and/or elastin have been associated with arterial stiffening in aging and cardiovascular disease [10, 12-16].
Cells sense changes in the stiffness of their microenvironment though a process called mechanotransduction, which typically involves the binding of ECM components to cell surface receptors and initiation of receptor-mediated signaling. Fibrillar collagens can bind to the discoidin domain receptor tyrosine kinases [17], but most studies have focused on the binding of fibrillar collagens to their receptors within the integrin family [18]. Integrin engagement leads to the recruitment of numerous signaling proteins and results in organization of the actin cytoskeleton, cellular stiffening and stiffness-mediated gene transcription [19-22]. The Rho and Rac non- receptor GTPases are among the major intracellular signaling components regulated by changes in ECM stiffness. In turn, the stiffness-dependent activation of these GTPases has profound effects on the actin cytoskeleton, migration, proliferation and differentiation [23-32]. The third non- receptor GTPase, Cdc42, also affects actin organization and motility [23- 33], but its role in SMC proliferation and differentiation is unclear.
We previously reported that a stiffness-dependent activation of Rac, but not Rho, promotes SMC proliferation in vitro by stimulating G1 phase cyclin D1 induction and S phase entry [9,34]. Additionally, genetic depletion of SMC Rac1 in vivo strongly reduces cyclin D1 expression, proliferation, and neointimal formation after femoral artery injury [34]. However, the stiffness- dependent activation of both Rac and Rho can regulate SMC differentiation [29], which could then lead to changes in ECM production and arterial stiffness as described above. This scenario raised the possibility that the reduced SMC proliferation observed in injured Rac1-deficient arteries [34] might be secondary to potential effects on SMC differentiation. Since the effect of Rac1 deficiency alone on in vivo SMC differentiation is not known, we examined the levels of SMC differentiation markers, performed functional tests of arterial contractility, and assessed the abundance of the major arterial collagens as well as elastin integrity in carotid artery sections of mice deficient for SMC Rac1. We also used pressure myography to compare carotid artery mechanics and contraction in the presence and absence of SMC Rac1.
RESULTS
Smooth muscle-specific deletion of Rac1
We generated floxed Rac1 mice harboring a SMC-specific, tamoxifen-inducible Cre transgene (hereafter called Rac1ff;iCre mice) and treated the mice with vehicle (ethanol) or tamoxifen. Consistent with our previous work [34], Rac1 was strongly expressed in the carotid artery medial layer of Rac1ff;iCre mice treated with vehicle and much reduced in the carotid medial layers of mice that had received tamoxifen [Figure 1A-B]. We note an unexpected tamoxifen effect on adventitial Rac1, but the levels of Rac1 in the adventitia were low as compared to those in the media [Figure 1A-B].
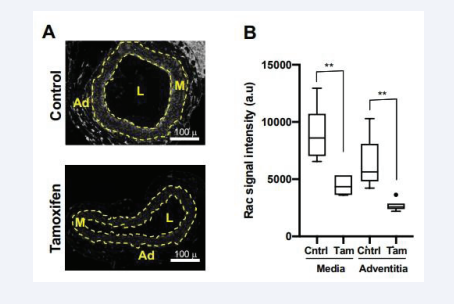
Figure 1: Deletion of smooth muscle Rac1. (A) Images of carotid artery cross-sections from Rac1f/f ;iCre mice that had been treated with ethanol (vehicle; control) or tamoxifen. The sections were immunostained with an antibody to Rac1. Scale bars = 100 μm. The dashed lines in panel A approximate the positions of the external and internal elastic laminae, which separate the medial (M) and adventitial (Ad) layers. The lumen is indicated as “L”. (B) Immunostaining results, as shown in A, were quantified and graphed as box plots with Tukey whiskers; n = 6 per condition. Statistical significance was determined by Mann–Whitney tests, comparing control versus tamoxifen treatment within the medial and adventitial layers.
Carotid artery morphology, as determined by H&E staining, appeared normal in both the vehicle and tamoxifen- treated mice [Figure S1, top panels].
SMC differentiation markers and arterial contraction
SM-MHC and α-SMA are among the best-established molecular markers of the differentiated SMC phenotype (see Introduction). We therefore used immunochemistry to examine the levels of these proteins in the medial layer of mouse carotid sections containing and lacking Rac1. The expression of medial SM-MHC trended lower in the carotid arteries of the Rac1- deficient mice, but the change was not statistically significant [Figure 2A-B].
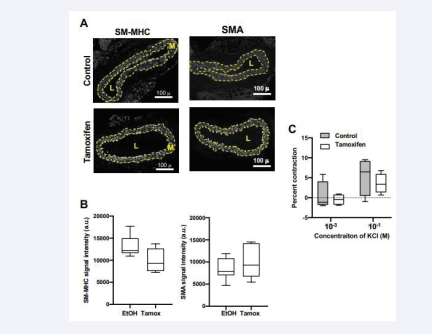
Figure 2: Minimal effect of in vivo Rac1 deletion on smooth muscle differentiation. (A) Representative images of carotid artery cross-sections from Rac1f/f ; iCre mice treated with ethanol (vehicle; control) or tamoxifen. The sections were immunostained with an antibody to SM-MHC or a-SMA. Dashed lines approximate the positions of the external and internal elastic laminae, which encompass the medial layer (M). The lumen is indicated as “L”. Scale bars = 100 μm. (B) Immunostaining results for medial SM-MHC (n=6) and medial a-SMA (n=7) were quantified and graphed as box plots with Tukey whiskers. Statistical significance was tested using Mann–Whitney tests. (C) Contraction to KCl in isolated carotid arteries from mice containing Rac1 (control; n=4) or deficient in Rac1 (tamoxifen; n=4). To exclude potential effects of tamoxifen beyond activation of the Cre transgene, the contraction graph for the control mice show combined results from Rac1f/f ; iCre mice that had been treated with ethanol (vehicle; n=2) and Rac1f/f mice (lacking the iCre transgene) that had been treated with tamoxifen (n=2). The contraction seen in response to 10-4 M KCl (see Methods) was set to zero (dashed line in panel C) as this KCl concentration approximated that of the HBSS buffer. Statistical significance in C was tested using ANOVA, comparing contraction in the control vs. tamoxifen treated samples for each KCl concentration.
The levels of medial α- SMA were not affected by Rac1 deficiency [Figure 2A-B]. We then extended these marker studies by examining the effect of SMC Rac1 deletion on arterial contraction, a key functional test of the contractile SMC phenotype. Contraction to KCl was not statistically different in the control and Rac1-deficient carotids [Figure. 2C]. Thus, despite its inhibitory effect on SMC proliferation after injury [34], in vivo deletion of Rac1 is not sufficient to alter SMC differentiation significantly as determined by both molecular marker analysis and functional testing of contractility.
Arterial ECM proteins and mechanics
Changes in SMC differentiation are also associated with remodeling of the arterial ECM, with increased synthesis of fibrillar collagens, degradation of elastin, and arterial stiffening being characteristic of the de-differentiated phenotype (see Introduction). These changes are thought to contribute to the increased proliferative potential of de-differentiated SMCs after injury and in atherosclerosis [9,10,13,14]. When we compared levels of the three major arterial fibrillar collagens (I, III and IV) in vehicle- and tamoxifen-treated carotid arteries, we found that the levels of these proteins were not significantly affected by the tamoxifen-mediated depletion of Rac1 [Figure 3A-B]. Similarly, preferential localization of collagen-I to the adventitia was seen in both the Rac- containing and Rac1-deficient carotid arteries [Figure 3A-B].
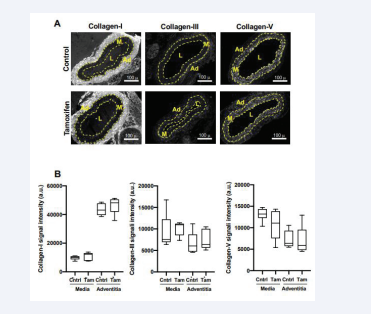
Figure 3: Arterial fibrillar collagen abundance is not affected by deletion of smooth muscle Rac1. (A) Images of carotid artery cross-sections from Rac1f/f ; iCre mice that had been treated with ethanol (vehicle; control) or tamoxifen. The sections were stained with antibodies to collagen-I, collagen-III, or collagen-V. Scale bars = 100 μm. Dashed lines approximate the positions of the external and internal elastic laminae, which separate the medial (M) and adventitial (Ad) layers. The lumen is indicated as “L”. (B) Immunostaining results, as shown in A, were quantified and graphed as box plots with Tukey whiskers; n=6 per condition. Statistically significant differences were not detected when comparing (by Mann–Whitney tests) control versus tamoxifen treatment within the medial and adventitial layers.
Collagen-III and -V were found in both the media and adventitia, and this expression pattern was, again, unaffected by Rac1 deficiency [Figure 3A-B]. Elastin integrity was also unaffected by Rac1 depletion [Figure S1, bottom panels].
Finally, we extended these targeted ECM studies with a functional approach in an effort to reveal potential effects of Rac1 on arterial stiffness that elude detection through the analysis of individual ECM components. To accomplish this goal, we performed biaxial inflation-extension tests of vehicle- and tamoxifen-treated mouse carotid arteries on a pressure myograph. This functional approach assesses the effects of Rac1 deletion on arterial mechanical responses to increasing pressure [35]. The results showed that metrics of carotid artery stiffness in the axial direction, as determined by the “in vivo stretch” (IVS; see Methods) and the axial stretch-strain relationship, were not affected by the deletion of SMC Rac1 [Figures 4A-B and S2].
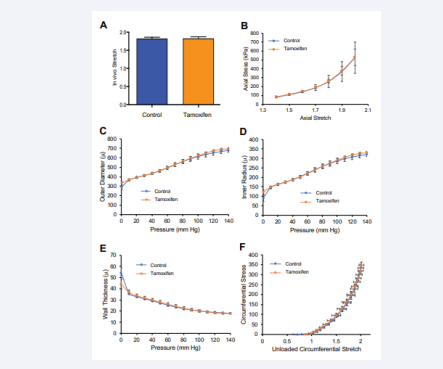
Figure 4: Deletion of SMC Rac1 does not affect arterial stiffness as determined by passive arterial mechanics. Freshly isolated carotid arteries from control mice (n=6) or Rac1f/f;iCre mice treated with tamoxifen (n=7) were analyzed by pressure myography. To exclude potential effects of tamoxifen beyond activation of the Cre transgene, graphs for the control mice show combined results from Rac1f/f iCre mice that had been treated with ethanol (vehicle; n=2) and Rac1ff mice (lacking the iCre transgene) that had been treated with tamoxifen (n=4). (A) In vitro stretch as determined from axial force-length tests. (B) Axial stress–stretch curves obtained at 80 mm Hg. (C) Changes in outer diameter with increasing pressure. (D and E) Changes in vessel inner radius and wall thickness, respectively, with pressure. (F) Circumferential stress–stretch curves with data points showing results at 0–140 mm Hg in increments of 10 mm. Results in panels A-B show means + SD, and results in panels C-F show means ± SE (see Methods). Statistical significance was determined by a MannWhitney test (panel A) or two-way ANOVAs (panels B-F) comparing control to tamoxifen. In F, statistical significance was tested for both stretch and stress.
Similarly, pressure-induced changes in arterial outer diameter [Figure 4C], inner radius [Figure 4D], wall thickness [Figure 4E], and arterial stiffness in the circumferential direction (as determined by absence of right or left shifts in the stretch-strain curves; [Figure 4F]) were not distinguishable in the carotid arteries containing or deficient for Rac1. Thus, depletion of SMC Rac1 did not affect either molecular markers or functional tests of arterial stiffness.
DISCUSSION
Changes in SMC differentiation have typically been linked to changes in Rho activity because Rho activation promotes cytoplasmic-nuclear shutting of transcriptions factors that regulate SMC differentiation genes such as Myh11, the gene encoding SM-MHC (reviewed in [36]). However, Talwar et al. [29], reported that Rac acts together with Rho to maintain SMCs in the contractile (differentiated) state. We therefore wondered if arterial depletion of Rac1 (but not Rho) might be sufficient to affect SMC differentiation and its consequent effects on ECM remodeling and arterial stiffness.
Our results show that Rac1-containing and -deficient carotid arteries similarly express SMC differentiation markers and do not show a statistically significant difference in their contractile response to KCl. Moreover, the abundance of the major arterial fibrillar collagens, the integrity of medial elastin, and several functional tests of arterial stiffness (as determined by pressure myography) were similar whether Rac1 was present or depleted from SMCs genetically. Taken together, we conclude that in vivo Rac1 deficiency in itself does not alter the basal, contractile state of arterial SMCs. These findings are notably different from those we recently described in carotid arteries deficient for Focal Adhesion Kinase (FAK), as medial FAK deletion was sufficient to reduce arterial contraction to KCl [37]. FAK activity is upstream of Rac-GTP activation [9,34], but FAK has many effectors beyond Rac1 (reviewed in [38]): one or more these other effectors must be required for regulation of SMC differentiation.
The data presented here also inform our understanding the stimulatory Rac1 effect on SMC proliferation. For example, if Rac1 were required for SMC de-differentiation, then in vivo Rac1 deficiency could have reduced injury-induced SMC proliferation [34] through an indirect inhibitory effect on SMC de-differentiation. Our results argue that this is not the case. While we cannot formally exclude potentially distinct effects of Rac1 in different arterial beds (femoral [9, 34] and carotid; this report), our combined results support the idea that the reduced in vivo SMC proliferation of arteries deficient for Rac1 reflects the stimulatory effect of Rac1 signaling to the cell cycle rather than being a secondary consequence of altered SMC differentiation or arterial stiffness.
METHODS
Mice and Artery Isolation
Mice on the C57BL/6 background harboring floxed Rac1 genes and a tamoxifen-inducible, SMC-specific Cre transgene were generated as described [34]. Since the SMC-specific Cre transgene is on the Y-chromosome [39], all experiments were performed on male mice. The mice were fed a chow diet ad libitum, aged to 3-4 months, treated with ethanol (vehicle) or tamoxifen (Sigma Aldrich) for 5 days as described [34], allowed to recover for 1–2 weeks, and sacrificed by CO2 asphyxiation at 4-5 months. The right carotid artery was immediately removed, stripped of most fat, and used for pressure myography. The left carotid artery was perfused in situ with PBS, removed, cleaned in PBS and fixed in Prefer (Anatech Ltd.) for paraffin-embedding, sectioning and immunostaining. Animal protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Pressure Myography
Arterial mechanics and the contractile response to KCl were determined ex vivo on a DMT 114P pressure myography with force transducer largely as described [37,40,41]. Isolated right carotid arteries (see above) were secured to 380 µm (outer diameter) cannulas using silk sutures, and submerged in 5-ml of calcium-containing Hanks Balanced Salt Solution (HBSS; Corning Life Sciences). This closed system was checked for leaks by pressurizing the vessel to 30-mm Hg using HBSS and ensuring that there was no pressure loss through the system. The arteries were also visualized by light microscopy, and the axial length (where the artery transitioned from being bent to straight) was recorded as the unstretched vessel length (UVL). Arteries were then brought to a stretch of 1.8 and pressurized to 100 mm Hg for 15 min with calcium-containing HBSS. The arteries were preconditioned by pressurization three times from 0–140 mm Hg. Unloaded (unstretched and unpressurized) vessel wall thickness and outer diameter were measured in multiple sections after preconditioning and averaged with measurements after axial testing for post-test data analysis. In vivo stretch (IVS) was determined using force-length tests as described [37,40], and the intersection of the force-stretch curves was defined as the IVS. Loaded inner radius and wall thickness were determined from pressure-outer diameter tests with samples at their IVS and pressurized in 10-mm Hg steps from 0-140 mm Hg (30-sec per step) before returning the artery to 0 mm Hg. The validity of IVS determinations was assessed by measuring axial force through the circumferential tests; we excluded samples where axial force varied from the mean by >25% with pressure. Raw measurements of the intraluminal pressure, force transducer readings, and video-tracked outer diameters were converted into stress-stretch curves as described [37,40]. Vessel wall thickness was calculated in the post-test analysis based on the sample being incompressible [40].
To measure the contractile response to KCl, freshly isolated left carotid arteries were cleaned of excess fat, mounted on the myograph, and incubated in 5 ml of HBSS as described above. The closed system was checked for leaks, returned to 0 mm Hg, and the UVL was determined, also as described above. Mounted vessels were preconditioned by stretching them axially to 1.15 times their UVL at 40 mm Hg for 15 min and then 1.3 times their UVL at 60 mm Hg for 15 min. Vessels were then brought to their IVS (approximately 1.8) and pressurized to 100 mm Hg. The vessel was allowed to equilibrate for 5 min. After baseline diameter measurements were taken, increasing concentrations of KCl (10-4 M, 10-3 M and 10-1 M in 5 ml HBSS) were added to the bath sequentially by first removing the preceding bath and adding the new dilution. Each dilution was allowed to reach a constriction plateau (~5-10 min) before outer diameter measurements were recorded and the next dilution added. KCl was rinsed out with HBSS until >95% of the original outer diameter was attained. Outer diameters were recorded in real time at each KCl concentration using MyoVIEW software (DMT) and an inline tracking camera (Imaging Source).
Immunofluorescence staining
Paraffin sections of isolated carotid arteries were prepared and immunostained for collagen-I, collagen-III, collagen-V, α-SMA, and SM-MHC as described [37,41,42]. Rac immunostaining used anti-Rac1 (catalog # 610650; BD Transduction Laboratories), typically with a ~100-fold primary antibody dilution and an Alexa Fluor secondary antibody (ThermoFisher). Results were visualized with a Nikon Eclipse 80i microscope with a QI- Click Qimaging camera. Carotid arteries were imaged at 20 x magnification. Images were quantified using Fiji. The media of each section was traced using the polygon drawing tool. Raw integrated intensity was divided by the area of the outlined media to yield values for relative fluorescence intensity. This procedure was repeated for the adventitial layer.
Statistical Analysis
The statistical tests were performed using Prism software (GraphPad). For the pressure myography experiments, in vivo axial stretch and the axial stress-stretch curves were determined from single measurements; graphs show mean ± SD. Pressure- mediated changes in outer diameter, inner radius, wall thickness,and circumferential stretch-stress results were determined from multiple measurements (see [40]); graphs show mean ± SE. Two- tailed Mann-Whitney tests were used to compare two datasets, and 2-way ANOVAs were used for multiple comparisons. Statistical significance for all graphs is demarcated by *(p<0.05),**(p<0.01), ***(p<0.001). Box plots show Tukey whiskers.
ACKNOWLEDGEMENTS
We thank V. Tybulewicz (National Institute of Medical Research, London) for the floxed Rac1 mice, and S. Offermanns (Max Planck Institute for Heart and Lung Research) for the smooth muscle–specific, inducible Cre mouse. This work was supported by NIH grant HL137232 to RKA.
REFERENCES
- Shirwany NA, Zou M. Arterial stiffness: a brief review. Acta Pharmacol Sin. 2010; 31: 1267-1276.
- Kohn JC, Lampi MC, Reinhart-King CA. Age-related vascular stiffening: Causes and consequences. Front Genet. 2015; 6: 112.
- Saphirstein RJ, Morgan KG. The Contribution of Vascular Smooth Muscle to Aortic Stiffness Across Length Scales. Microcirculation. 2014; 21: 201–207.
- Majesky MW, Dong XR, Hoglund V, Mahoney WM, Daum G. The adventitia: a dynamic interface containing resident progenitor cells. Arterioscler Thromb Vasc Biol.2011; 31:1530–1539
- Thyberg J, Blomgren K, Roy J, Tran PK, Hedin U.Phenotypic modulation of smooth muscle cells after arterial injury is associated with changes in the distribution of laminin and fibronectin.J Histochem Cytochem. 1997; 45: 837–846.
- Thyberg J, Hedin U, Sjolund M, Palmberg L, Bottger BA. Regulation of differentiated properties and proliferation of arterial smooth muscle cells. Arteriosclerosis. 1990; 10: 966-990.
- Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev.1995; 75: 487-517.
- Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res.2005; 96: 280-291.
- Klein EA, Yin L, Kothapalli D, Castagnino P, Byfield FJ, Xu T, et al. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr Biol. 2009; 19: 1511-1518.
- Kothapalli D, Liu S-L, Bae YH, Monslow J,Xu T, Hawthorne EA, et al. Cardiovascular Protection by ApoE and ApoE- HDL Linked to Suppression of ECM Gene Expression and Arterial Stiffening. Cell Rep. 2012; 2:1259-1271.
- Razinia Z, Castagnino P, Xu T, Vázquez-Salgado A, Puré E, Assoian RK. Stiffness-dependent motility and proliferation uncoupled by deletion of CD44. Sci Rep. 2017; 7: 16499.
- Thyberg J. Phenotypic modulation of smooth muscle cells during formation of neointimal thickenings following vascular injury. Histol Histopathol. 1998; 13: 871-891.
- Liu SL, Bae YH, Yu C,Monslow J, Hawthorne EA, Castagnino P, et al. Matrix metalloproteinase-12 is an essential mediator of acute and chronic arterial stiffening. Sci Rep. 2015; 5: 17189.
- Liu S-L, Bajpai A, Hawthorne EA, Bea Y, Castagnino P, Manslow J, et al. Cardiovascular protection in females linked to estrogen- dependent inhibition of arterial stiffening and macrophage MMP12. JCI Insight. 2019; 4: e122742.
- Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008; 75: 346-359.
- Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011; 4: 165-178.
- Carafoli F, Hohenester E. Collagen recognition and transmembrane signalling by discoidin domain receptors. Biochim Biophys Acta. 2013; 1834: 2187–2194.
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002; 110: 673–687.
- Sastry SK, Burridge K. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exp Cell Res. 2000; 261: 25–36.
- Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol. 2001; 13: 584–592.
- Wolfenson H, Yang B, Sheetz MP. Steps in Mechanotransduction Pathways that Control Cell Morphology. Annu Rev Physiol. 2019; 81: 585–605.
- Burridge K, Guilluy C. Focal adhesions, stress fibers and mechanical tension. Exp Cell Res. 2016; 343: 14–20.
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005; 33: 891–895.
- Lawson CD, Burridge K. The on-off relationship of Rho and Rac during integrin- mediated adhesion and cell migration. Small GTPases. 2014; 5: e27958.
- Provenzano PP, Keely PJ. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J Cell Sci. 2011; 124: 1195–1205.
- Pasapera AM, Plotnikov S V, Fischer RS, Case LB, Egelhoff TT, Waterman CM. Rac1-Dependent Phosphorylation and Focal Adhesion Recruitment of Myosin IIA Regulates Migration and Mechanosensing. Curr Biol. 2015; 25: 175-186.
- Avalos AM, Arthur WT, Schneider P, Quest AF, Burridge K, Leyton L. Aggregation of integrins and RhoA activation are required for Thy- 1-induced morphological changes in astrocytes. J Biol Chem. 2004; 279: 39139–39145.
- Lessey EC, Guilluy C, Burridge K. From mechanical force to RhoA activation. Biochemistry. 2012; 51: 7420-7432.
- Talwar S, Kant A, Xu T, Shenoy VB, Assoian RK. Mechanosensitive smooth muscle cell phenotypic plasticity emerging from a null state and the balance between Rac and Rho. Cell Rep. 2021; 35: 109019.
- Assoian RK, Klein EA. Growth control by intracellular tension and extracellular stiffness. Trends Cell Biol. 2008; 18: 347-352.
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005; 8:241-254.
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004; 265:23-32.
- Cerione RA. Cdc42: new roads to travel. Trends Cell Biol. 2004; 14:127-132.
- Bae YH, Mui KL, Hsu BY, Liu S-L, Cretu A, Razinia Z, at el. A FAK-Cas- Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci Signal. 2014; 7: ra57.
- Ferruzzi J, Bersi MR, Humphrey JD. Biomechanical phenotyping of central arteries in health and disease: Advantages of and methods for murine models. Ann Biomed Eng. 2013; 41: 1311-1330.
- Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010; 11: 353-365.
- Roberts E, Xu T, Assoian R. Cell contractility and Focal Adhesion Kinase control circumferential arterial stiffness. Vasc Biol. 2022; 4: 28-39.
- Guan JL. Focal adhesion kinase in integrin signaling. Matrix Biol. 1997; 16: 195-200.
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008; 14: 64-68.
- Brankovic SA, Hawthorne EA, Yu X, Zhang Y, Assoian RK. MMP12 Deletion Preferentially Attenuates Axial Stiffening of Aging Arteries. J Biomech Eng. 2019; 141: 0810041- 0810049.
- Kleeck RV, Roberts E, Castagnino P, Bruun K, Brankovic SA, Hawthorne EA, et al. Arterial stiffness and cardiac dysfunction in Hutchinson– Gilford Progeria Syndrome corrected by inhibition of lysyl oxidase. Life Sci Alliance. 2021; 4: e202000997.
- von Kleeck R, Castagnino P, Roberts E, Talwar S, Ferrari G, Assoian RK. Decreased vascular smooth muscle contractility in Hutchinson– Gilford Progeria Syndrome linked to defective smooth muscle myosin heavy chain expression. Sci Rep. 2021; 11: 10625.









































































































































{kind=link}