Role of Caspase-8 on Retinal Endothelial Cell Migration and Angiogenesis
- 1. Department of Molecular Science, The University of Texas Rio Grande Valley - School of Medicine, USA
ABSTRACT
Pathological angiogenesis is the hallmark of proliferative diabetic retinopathy (DR). In response to prolonged tissue injury, human retinal microvascular endothelial (HRMEC) cells form new blood vessels to ensure the supply of oxygen and nutrients; however, progressive fibrovascular proliferation can eventually lead to angiogenesis, retinal detachment, and blindness. Caspase-8, a responsible protease for apoptosis has also been shown to modulate pathological and developmental angiogenesis but whether it exerts this role in a hyperglycemic environment has not yet been explored. Here we demonstrate that high glucose decreased HRMEC cell viability and increased VEGF secretion. Pro-Caspase 8 expression was measured by RT-PCR and caspase 8 enzyme activity was confirmed by Caspase-Glo assay. In addition, Caspase-8 KD resulted in an inhibition of cell migration, and a significant reduction of p38 phosphorylation. Taken together, our data suggest that Caspase-8 may play a role in angiogenesis by promoting HRMEC migration via p38 MAPK phosphorylation.
KEYWORDS
- Caspase-8
- Retinal endothelial cells
- Angiogenesis
- p38
- Cell migration
CITATION
Campano P, Valdez L, Guerra L, Cheng B, Tsin A (2023) Resistant Hypertension in Thoracoabdominal Aortic Dissection Involving the Renal Arteries. Ann Vasc Med Res 10(1): 1154.
ABBREVIATIONS
DR: Diabetic Retinopathy; HMREC: Human Retinal Microvascular Endothelial Cells; DM: Diabetes Mellitus; T1D: Type 1 Diabetes; T2D: Type 2 Diabetes; PDR: Proliferative Diabetic Retinopathy; DISC: Death-Inducing Signaling Complex; FLIP: FLICE Like Interleukin 1ß-Converting Enzyme; MLKL: Mixed-Lineage Kinase Domain-Like; RIPK3: serine/threonine protein kinase; VEGF: Vascular Endothelial Growth Factor Cells; HIF-1: Hypoxia-Inducible Factor-1; sICAM-1: Soluble Intercellular Adhesion Molecule-1; MAPK: P38-Mitogen Activated Protein Kinase
INTRODUCTION
Diabetic retinopathy (DR) is a condition affecting individuals with prolonged diabetes mellitus (DM) that is characterized by lesions in the retina. Not only is DR one of the most severe consequences arising from DM, but it is also a leading cause of blindness in the world [1]. Type 2 diabetes (T2D) has reached an epidemic level globally while type 1 diabetes (T1D) rates are rapidly rising [2]. In the United States, the prevalence rate of diabetes in Hispanics is almost twice that of Caucasians. Moreover, 48% of the individuals aged ≥40 in the Hispanic community suffer from DR with 32% suffering from moderate to severe non-proliferative and proliferative retinopathy [3].
The retina is one of the most metabolically active tissues in the body, consuming high levels of oxygen and nutrients. The microvasculature in the retina responds to hyperglycemia through a number of biochemical changes that can be divided into an early and late stage. In the early non-proliferative stage, DR is denoted by pericyte dropout that leads to microaneurysms, thickening of the basement membrane, and increased permeability [4]. These events are preceded by ischemia of the retinal tissue which promotes upregulation of endothelial mitogens for secretion of growth factors. Extensive fibrovascular proliferations may lead to retinal detachment that is sometimes accompanied by vitreous hemorrhage. At the end stage of DR, when the vitreous has been completely contracted, loss of vision is best explained by severe retinal ischemia [5]. Because non-proliferative DR can progress to proliferative diabetic retinopathy (PDR), it is important to understand the biological processes that govern retinal vascular development. This has become an area of interest for researchers and clinicians to develop preventive and interventional therapeutics for vascular eye diseases that address promoters of abnormal vascular growth.
The protein of interest in this project is caspase-8, a cysteine- aspartate specific protease that is well known for activation of the extrinsic cell death pathway [6-9]. Apoptosis occurs when procaspase-8 is recruited by its N-terminal prodomain to the death-inducing signaling complex (DISC) following a death receptor ligation [6,9]. Caspase-8 aggregate and facilitate their dimerization of the C-terminal catalytic domain through the proximity-induced model which initiates autocatalytic processing [6,7,9]. The released, active dimer cleaves a variety of downstream targets. However, caspase-8 activity does not always result in cell death. In fact, low levels of caspase-8 activity promote the formation of the ripoptosome complex in which caspase-8 forms a heterodimer complex with FLIP. The FLICE like Interleukin 1ß-Converting Enzyme (FLIP) is a catalytically inactive homologue of caspase-8 and is also recruited during the DISC formation. Among the caspase family, caspase-8 is unique in that it acts as an environmental sensor, transduces a range of signals to cells, and modulates responses that extend beyond survival [10]. While it is well known for its role in cell-death signaling, caspase-8 has also been shown to take part in activities that promote cell survival independently of its cell-death processes which include cell migration and survival [6,11-13].
There are many molecular mechanisms that are the causes of vascular dysfunction [14]. For example, hyperglycemic induced hypoxia results in the secretion of vascular endothelial growth factor (VEGF) from the endothelial cells within the retina [15]. VEGF also acts as a modulator of blood vessel growth and strongly increases vascular permeability. While VEGF is primarily used for survival factor for endothelial cells, it also can function by stimulating proliferation, migration, and tube formation. In DR, VEGF is excessively produced and results in endothelial cell proliferation, tissue damage, fluid leakage, new blood vessel formation and microaneurysms, playing a major role in disease progression. Growth factors such as VEGF promote a neovascular response locally and by diffusing through the vitreous to other areas of the retina, to the optic disc, and into the anterior chamber.
P38-mitogen activated protein kinase (MAPK) cascades are known as signaling pathways involved in regulating endothelial cells. Endothelial cells are regulated in response to external and internal stimuli such as growth factors and stress. The p38 signaling pathway also controls apoptosis, as well as the release of cytokines from neutrophils and macrophages when activated. The p38 pathway is activated by environmental stress and inflammatory cytokines and to play a part in the inflammation process in many diseases. MAPK during neutrophil apoptosis has been shown to associate with caspase 8. P38-MAPK activity has also been shown to be a survival signal in these primary cells.
MATERIALS AND METHODS
Cell Culture and glucose exposure
Primary Human Retinal Microvascular Endothelial Cells (HRMEC) were purchased at passage 3 from Cell Systems, Kirkland, WA and propagated using Complete Classic Medium, Gibco™ Antibiotic- Antimycotic, and Culture Boost™. All cells used in the experiments were either passage 5 or 6 and were incubated at 37°C at 5% CO2. Cells used to measure glucose exposure were supplemented with physiological (5.5mM) or hyperglycemic (30mM) glucose levels using glucose-free DMEM prepared with 1% Gibco™ Antibiotic-Antimycotic and 10% FBS. Viable cells were counted using the Trypan blue dye exclusion method.
Real-time polymerase chain reaction
HRMEC were seeded into a 6 well plate at a density of 8 x 104 and treated with 5.5mM or 30mM glucose. Cell lysates were collected after 24 hours and stored at -80°C. One microgram total RNA was reversed transcribed with SuperScript™. CTATCCTGTTCTCTTGGAGAGTCC-3’; Caspase-8: forward primer, 5’ CACTAGAAAGGAGGAGATGGAAAG-3’; reverse primer, and 18S which was used as the housekeeping gene, forward primer; 5’ CGGAGTCAAGGATTTGGTCGTAT-3’, 5’- AGCCTTCTCCATGGTGGTAAGA-3’; reverse primer. Polymerase chain reactions were run on Gene Amp PCR system 9700 (Applied Biosystems, Foster City, CA) using a “hot start” method with the following conditions: 94 ºC for 5min, 94ºC for 1min, 60–65ºC for 1min, 72 ºC for 1min, 72ºC for 10min. RT-PCR was performed using PowerUp SYBR Green Master Mix (Thermo Scientific).
Human VEGF Immunoassay
Cells were cultured with prepared media as indicated in cell culture and ‘glucose exposure section’. Conditioned medium was harvested from cells and stored at -20oC overnight for ELISA. Human VEGF ELISA (Quantikine ELISA Catalog number: DVE00) was performed per manufacturer instructions and analyzed using a Biotek plate reader equipped with Gen5 microplate reader with manager software). Data was quantified in comparison to VEGF standards.
Casp-8 Glo assay
HRMEC were seeded at 3.5x104 in an opaque bottom 96-well tissue culture plate (ref: 353296, Falcon™). After 24 hours, ECs were treated with 5.5mM or 30mM of glucose. The Casp-8 Glo assay was performed 24 hours following treatment according to the manufacturer’s instructions (Promega) using a Thermo ScientificTM FlouroskanTM FL plate reader and Skan it 6.0.2 software.
siRNA Transfection
HRMEC were seeded in a 24 well plate at a density of 4.5x104. At 80% confluent, cells were transfected dropwise with validated Caspase-8 Silencer™ Select Pre-Designed siRNA® (6 RefSeqs; assay ID: s2427) and Silencer™ Negative Control No. 1 siRNA (Life Technologies) according to the manufacturer’s instructions. Two days following transfection, cells were treated with 5.5mM or 30mM of glucose. The final concentration of siRNA solution was 5pmol per well.
Immunoblotting
HMREC used for western bot analysis were plated at 1x106 in 100mm dishes and transfected with Caspase-8 Silencer™ Select Pre-Designed siRNA® or Silencer™ Negative Control No. 1. Final concentration of siRNA solution was 600pmol. Cells were lysed using RIPA lysis buffer mix. Protein from the cell lysates were separated by SDS-PAGE and immunoblotted according to standard protocols. 2%BSA was used for blocking p-p38. B-actin (1:10000, Lot:00923585, Invitrogen), p-p38 (1:1000, ref. 4511), Cell Signaling, and p38 (1:1000, ref. 9212, Cell Signaling).
Scratch Wound Assay
After transfected cells were confluent, they were starved in 2% FBS DMEM medium overnight. A wound was induced by scraping the cell monolayer with a P200 pipette tip. Photographs were acquired at time-point 0,12, and 24 hours after incubation at 37°C. Images were captured using Nikon fluorescence microscope at 10X. Images for scratch assay of 0, 12, and 24 hours were analyzed using ImageJ software version 1.53a. Wound area for each image was outlined and determined using the measuring tool.
Analysis of wound closure
Wound area for each image was outlined and determined using the measuring tool. 1 pixel represents 1 um2. Images were captured using Nikon fluorescence microscope at 10x. Images for scratch assay of 0, 12, and 24 hours were analyzed using ImageJ software version 1.53a.
Statistical Analyses
Students T-test was used for calculating statistical significance. Error bars represent Mean± SEM. (*p<0.01).
RESULTS AND DISCUSSION
Hyperglycemia reduces HRMEC viability
There were 200,000 viable cells at time of treatment. After 24 hours, HRMEC in 5.5mM glucose the viable cell number increased to 232,500 while cells in 30mM showed a decrease of 42% to 119,000 [Figure 1]. These findings are in line with previous studies showing that high glucose in endothelial cells may impact the integrity of the microvasculature in the diabetic retina leading to angiogenesis [15].
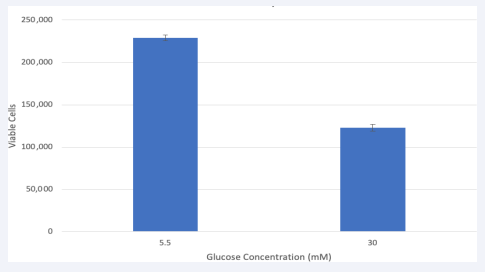
Figure 1 Hyperglycemia treatment decreased viable HRMEC.
Cells were seeded in a 24 well plate. At confluency, cells were treated, and viable cell number had increased to 200,000. After 24 hours of glucose treatment, there were 232,500 viable cells in 5.5mM and 119,000 in 30mM. Data is represented in Mean ± SEM. P=.01, T-test. N=3.
Hyperglycemia increases VEGF secretion
Figure 2 shows VEGF levels in cell medium after 24-hour glucose treatment. Compared to VEGF in cell media from the 5.5mM glucose group, VEGF levels increased when treated with 30mM of glucose from 16to 22pg/mL. This suggests that hyperglycemic cells were induced to release more VEGF. Results from a t-test show that the difference in VEGF secretion was significant (P<.01). The VEGF per cell (pg/cell) was calculated after 24 hours and each cell in the 5.5mM group secreted 0.000176pg/cell whereas the 30mM group secreted 0.000341pg/ cell. The increase in glucose concentration is consistent with increased VEGF secretion in HRMEC suggesting a pro-angiogenic response as it occurs in the diabetic retina.
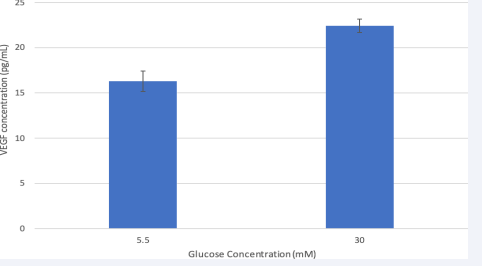
Figure 2 Hyperglycemia increased VEGF level in the conditioned media of HMREC.
After 24-hour glucose treatment, VEGF protein in cell media was measured for by ELISA. Data is represented in Mean ± SEM. Results from a t-test were significant. P=0.01, T-test. N=3
Pro-Caspase-8 gene expression in HRMEC
Next, we investigated whether hyperglycemia would influence caspase-8 expression in HRMEC. RT- PCR analysis revealed that cells treated with high glucose increased in pro- caspase-8 mRNA compared to the physiological group [Figure 3]. It has been shown that high glucose mediates human pancreatic cell apoptosis via up-regulation of the Fas receptor as well as in human coronary artery endothelial cells [16,17]. However, whether the increase in expression explains the cell death earlier observed in hyperglycemic HRMEC is unknown because caspase-8 is initially synthesized as a single-chain zymogen (procaspase-8) [9]. Caspase-8 are recruited to the DISC as intact monomers and this recruitment subsequently leads to their dimerization and activation through induced proximity [8].
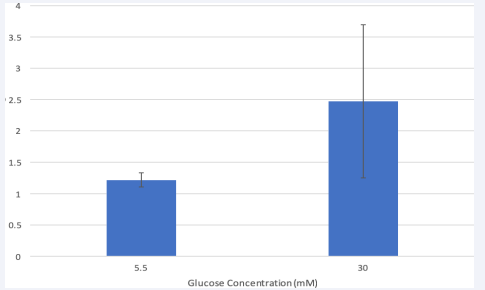
Figure 3 Hyperglycemia increased pro-caspase-8 expression in HMREC.
Pro-caspase-8 mRNA was measured by quantitative real time PCR after 24- hour treatment. Procaspase-8 at indicated glucose concentrations were normalized according to the expression of housekeeping gene 18s as a control. Data is represented in Mean ± SEM. P= 0.36, T-test. N=3
HRMEC exhibits Caspase-8 Activity
Because Caspase-8 activity is required to promote proper cell migration [18], a Caspase-8 Glo assay was conducted. HRMEC in physiological glucose levels showed a slightly lower caspase-8 activity than hyperglycemic cells [Figure 4]. The increase of caspase-8 activity is consistent with increase of VEGF protein previously shown by the ELISA. Both glucose groups showed between 1.5 and 2 luminescence units, falling at the lower end of the standard scale. Our Glo-Assay provides us with further insight that could explain what occurs at the DISC complex once procaspases are recruited. Studies imply that the Caspase-8/ FLIP heterodimer with lower activity levels is likely to be responsible for cell migration [18] and is not the cause of the fully mature caspase-8 homodimer promoting the extrinsic cell death pathway [18]. Furthermore, Maedler et al., [16] found that up-regulation of FLIP, by incubation with transforming growth factor or by transfection with an expression vector coding for FLIP, protected β cells from glucose induced apoptosis, restored β cell proliferation, and improved β cell function.
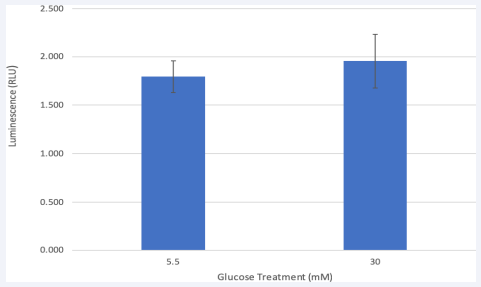
Figure 4 Caspase-8 enzyme activity in HRMEC.
Caspase-8 Glo assay shows that Caspase-8 activity in the euglycemic HRMEC is slightly lower after 24-hr. treatment. Normal and high glucose groups both associated with a low level of Caspase-8 activity. Activity of Caspase-8 is proportional to luminescent signal which is measured in relative light units. Data is represented in Mean ± SEM. P=0.66, T-test. N=3.
Requirement of Caspase-8 for HRMEC migration
After confirming caspase-8 activity in HRMEC, we studied if silencing Caspase-8 would prevent cell migration in a hyperglycemic setting. Prior to treating cells with glucose, an rt- PCR was performed to ensure the knockdown efficacy [Figure 5].
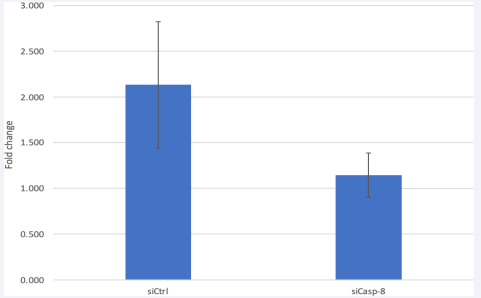
Figure 5 Caspase-8 KD reduces caspase 8 expression in HRMEC.
Caspase-8 mRNA was measured by RT-PCR after Si-RNA transfection. Quantitative real time PCR showed a lower expression in siCasp-8 vs the control. Data is represented in Mean ± SEM. P=0.24, T-test. N=3.
Following transfection, cells were treated with high and low glucose for 24 hours and images were taken at 0,12, and 24 hours to track wound closure [Figure 6]. The 5.5 mM glucose treated Casp-8 KD EC group wound area decreased before reversing to almost its original size at 24 hours. Analyzation with Image J software revealed a decreased surface area overall in the KD vs. the control group with no difference between the normal and high glucose group [Figure 7].

Figure 6 Requirement of Caspase-8 on HMREC migration.
Cell migration was based on wound closure/scratch assay. Representative bright field images of the scratch wound assay of HMREC transfected with control siRNA or Casp-8 and 5.5 or 30 mM of glucose are shown. Wound closure after 24hr of scratch is reduced in siCASP-8 compared to siCtrl.1 pixel represents 1um2. N=3.
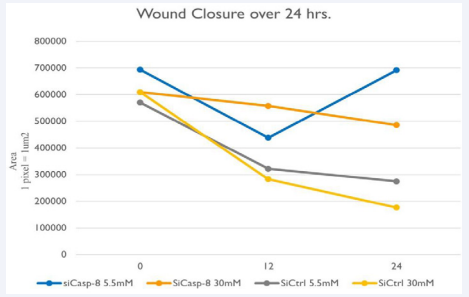
Figure 7 Wound Closure Analysis
Wound area measured where 1 pixel represents 1um2 of the different HRMEC groups at 0,12, and 24 hours. siCtrl groups show the greatest decrease in surface area (or increased cell migration) at 12 hours, and at 24 hours, suggesting that si-Casp-8 inhibited cell migration.
Requirement of Caspase-8 on p38 MAPK phosphorylation
The p38 mitogen-activated protein kinase (MAPK) cascades are signaling pathways that regulate multiple functions of endothelial cells in response to exogenous and endogenous stimuli such as growth factors, stressors, and cytokines. Loss of caspase-8 has been shown to increase basal p38 phosphorylation [18,19]. The proposed pathway is mediated by RIPK3 phosphorylation which results in increased downstream phosphorylation of p38 upon loss of caspase-8 [18]. This p-p38 activity is the presumed downstream effector responsible for preventing the proper response to VEGF in EC resulting in impaired angiogenesis [18]. Contrary to our expected results, western blot analysis revealed a decrease of p38 phosphorylated protein in the siCaspase-8 group compared to the control group [Figure 8]. Tish et al. [18], also demonstrates that stimulation of Casp-8 KD ECs with VEGF further increases p-p38. It is possible that stimulation of HRMEC with VEGF to increase its protein concentration, could have resulted in increased p38 MAPK.
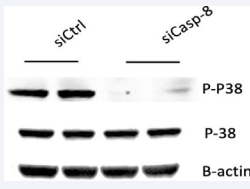
Figure 8 Requirement of Caspase-8 on p-38 MARK phosphorylation in HRMEC.
Western blot analysis shows that p38 signaling pathways are normally activated in basal conditions siCtrl and is decreased in si-Casp-8 cells.
DISCUSSION
Our studies have shown that hyperglycemia has a negative effect on endothelial cells by decreasing viability. Whether this is the result of Caspase-8 mediated apoptosis is unknown as cleaved Caspase-3 was not studied in this project. Identifying Fas-receptor activation in comparison to Caspase- 8/FLIP protein levels could have provided further insight as the direct effect glucose has on caspase-8. Because caspase-8 is initially synthesized as a single-chain zymogen (procaspase-8) [9] we could not rule out that caspase-8 was further processed to become a mature caspase-8. However, Tish et al. [18], showed that Casp- 8–mediated apoptosis may have played a small but significant contribution to the physiological remodeling process in HUVEC, without affecting overall vessel regression. Furthermore, it is possible that TGFβ mediated apoptosis played a role in the cell death seen in the hyperglycemic group. A study by Betts-Obregon 2016 showed that TGFβ significantly induced apoptosis in retinal pericytes and endothelial cells which also accumulated in the inner retina arterioles.
It was important for us to identify VEGF secretion in HRMEC as this is an endothelial cell mitogen and key regulator of both physiological and pathological angiogenesis [19,20]. Here we also demonstrate that hyperglycemic HRMEC release higher levels of VEGF protein in comparison to the physiological glucose level group supporting previous results showing concentration- dependent increase of VEGF protein in human retinal endothelial cells and pericytes [1,2]. It has become clear that Caspase-8 possesses more function than its long- established role in triggering apoptosis. Here we demonstrate that knockdown of caspase-8 inhibits cell migration and results in decreased p38 phosphorylation . In cancer cells, p38 regulates cell migration through hypoxia-induced activation of HIF-1 that leads to the expression of VEGF which binds to tyrosine kinase receptor VEGFR2 present at the surface of blood vessel endothelial cells. Downstream of p38, MAPKAP kinase-2 (MK2), contributes to increase in the level of polymerized actin by phosphorylating HSP27 leading to actin polymerization, focal adhesion, turnover, and cell contraction lead to cell migration. This pathway has been identified in a study of endothelial cells where RIPK3 positively regulated the activation of p38 and HSP27 upon permeability factor treatment (VEGF-A, VEGF-B, FGF-b) and thus, promoting permeability in the tumor cells to metastasize [19-25]. This potentially explains the loss of EC migration in HRMEC. In Tisch’s study, in the absence of Casp- 8, RIPK3 mediates increased phosphorylation of p38 in vitro and impaired developmental vascular growth in vivo. This RIPK3/p-p38 axis deregulates EC behavior, resulting in impaired angiogenesis [26-37]. This suggests that the loss of caspase-8 resulted in decreased p38 phosphorylation in HRMEC rather than the increase phosphorylation, may be specific to HRMEC. Further studies are needed to identify the molecular switches that govern caspase-8 behavior to stop from becoming active dimer in high glucose conditions to a caspase-8/FLIP heterodimer that influence cell migration. Repeated studies that test for Fas receptor activation and subsequently p38 phosphorylation in hyperglycemic Caspase-8KD EC could provide us with further insight on how high glucose affects EC behavior [37-39].
CONCLUSION
High glucose decreased HRMEC cell viability and increased VEGF secretion. Pro- Caspase 8 expression was detected by RT-PCR and Caspase-8 activity was measured by Caspase-Glo assay in HRMEC. Additionally, Caspase-8 KD resulted in an inhibition of cell migration, and a significant reduction of p38 phosphorylation. Our results suggest that Caspase-8 may play a role in DR angiogenesis by the promotion of MHREC migration and p38 MARK phosphorylation.
ACKNOWLEDGEMENTS
This research was supported by grants from the NIH (R15EY0033551), the Meadow Foundation and the Valley Baptist Legacy Foundation.
REFERENCES
- Bretts-Obregon BS, Buikema J, Tsin A, Vellanki S, Wright K. Effect of Glucose on Retinal Endothelial Cell Viability and VEGF Secretion. HSOA J Cell Biol Cell Metabol. 2016; 3:1–4.
- Duh EJ, Sun JK, Stitt AW. Diabetic retinopathy: current understanding,mechanisms, and treatment strategies. JCI insight. 2017; 2: 1–13.
- West SK, Klein R, Rodriguez J, Munoz B, Broman AT, Snyder R, et al.Diabetes and Diabetic Retinopathy in a Mexican American Population: Proyecto VER. Diabetes Care. 2001; 24: 1204-1209
- Al-Kharashi AS. Role of oxidative stress, inflammation, hypoxia and angiogenesis in the development of diabetic retinopathy. Saudi J Ophthalmology. 2018; 32: 318–323.
- Danis R, Davis MD. Proliferative diabetic retinopathy. ContemporaryDiabetes: Diabetic Retinopathy. 2012; 5: 969–1000.
- Mandal R, Barrón JC, Kostova I, Becker S, Strebhardt K. Caspase-8: Thedouble-edged sword. BBA – Reviews on Cancer. 2020; 1873: 188357.
- Van Raam BJ, Salvesen GS. Proliferative versus apoptotic functions of caspase-8: Hetero or homo: The caspase-8 dimer controls cell fate. Biochim Biophys Acta. 2012; 1824:113–122.
- Zhao Y, Sui X, Hong R. From procaspase-8 to caspase-8: Revisiting structural functions of caspase-8. J Cellular Physiology. 2010; 225: 316–320.
- Clauss M, Breier G. Mechanisms of angiogenesis. Springer Science &Business Media. 2004; 94: 1-71.
- Tae-Bong Kang, Wallach D, Ben-Moshe T, Varfolomeev EE, Pewzner- Jung Y, Yogev N, et al. Caspase-8 Serves Both Apoptotic and Nonapoptotic Roles. J Immunol. 2004; 173: 2976–2984.
- Chen S, Tisch N, Kegel M, Yerbes R, Hermann R, Hudalla H, et al. CNS Macrophages Control Neurovascular Development via CD95L. Cell Reports. 2017; 19: 1378–1393.
- Fritsch M, Gunther SD, Schwarzer R, Marie-Christine Albert, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019; 575, 683–687.
- Gui F, You Z, Fu S, Wu H, Zhang Y. Endothelial Dysfunction in DiabeticRetinopathy. Front Endocrinol (Lausanne). 2020; 11: 591.
- Betts-Obregon BS, Mondragon AA, Mendiola AS, Lebaron RG, Asmis R, Zou T, et al. TGFβ induces BIGH3 expression and human retinal pericyte apoptosis: A novel pathway of diabetic retinopathy. Eye. 2016; 30: 1639–1647.
- Maedler K, Fontana A, Ris F, Sergeev P, Toso C, Oberholzer J, et al. FLIP switches Fas-mediated glucose signaling in human pancreatic β cells from apoptosis to cell replication. Proc Natl Acad Sci U S A. 2002; 99: 8236-8241
- Kageyama SI, Yokoo H, Tomita K, Kageyama-Yahara N, Uchimido R, Matsuda N, et al. High glucose-induced apoptosis in human coronary artery endothelial cells involves up- regulation of death receptors. Cardiovasc Diabetol. 2011; 10: 73.
- Tisch N, Freire-Valls A, Yerbes R, Paredes I, Porta S L, Wang X, et al. Caspase-8 modulates physiological and pathological angiogenesis during retina development. J Clin Invest. 2019; 129: 5092–5107.
- Lee P, Lee DJ, Chan C, Chen SW, Chen I, Jamora C. Dynamic expressionof epidermal caspase 8 simulates a wound healing response. Nature.2009; 458: 519– 523.
- Hanggi K, Vasilikos L, Valls AF, Yerbes R, Knop J, Spilgies LM, et al. RIPK1/RIPK3 promotes vascular permeability to allow tumor cell extravasation independent of its necroptotic function. Cell Death Dis. 2017; 8: 1–11.
- Aiello LP, Wong JS. Role of vascular endothelial growth factor in diabetic vascular complications. Kidney Int Suppl. 2000; 58: 113–119.
- Albrecht EA, Sarma JV, Ward PA. Activation by C5a of endothelial cellcaspase 8 and cFLIP. Inflamm Res. 2009; 58: 30–37.
- Alvarado-Kristensson M, Melander F, Leandersson K, Ronnstrand L, Wernstedt C, Andersson T. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J Exp Med. 2004; 199: 449–458.
- Apte RS, Chen DS, Ferrara N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell. 2019; 176: 1248–1264.
- Bharadwaj AS, Appukuttan B, Wilmarth PA, Pan Y, Stempel AJ, Chipps TJ, et al. Role of the retinal endothelial cell in ocular disease. Prog Retin Eye Res. 2013; 32: 102–180.
- Crawford T, Alfaro III D, Kerrison J, Jablon E. Diabetic Retinopathy and Angiogenesis. Diabetes Reviews. 2009; 5: 8–13.
- Ferrara N. Vascular endothelial growth factor. Arteriosclerosis,thrombosis, and vascular biology. 2009; 29: 789–791.
- Forrester JV, Kuffova L, Delibegovic M. The Role of Inflammation in Diabetic Retinopathy. Front Immunol. 2020; 11: 583687.
- Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005; 438: 960–966.
- Guo H, Zhang L. EGR1/2 Inhibits Papillary Thyroid Carcinoma Cell Growth by Suppressing the Expression of PTEN and BAX. Biochem Genet. 2021; 59: 1544-1557.
- Grammas P, Riden M. Retinal endothelial cells are more susceptible to oxidative stress and increased permeability than brain-derived endothelial cells. Microvasc Res. 2003; 65: 18–23.
- Heimsath EG, Unda R, Vidro E, Muniz A, Villazana-Espinoza ET, Tsin A. ARPE- 19 cell growth and cell functions in euglycemic culture media. Curr Eye Res. 2006; 31: 1073–1080.
- Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, et al. Co- operative and Hierarchical Binding of c- FLIP and Caspase-8: A Unified Model.Defines How c-FLIP Isoforms Differentially Control Cell Fate. Molecular Cell. 2016; 61: 834–849.
- Jonkman JEN, Cathcart JA, Xu F, Bartolini ME, Amon JE, Stevens KM, et al. An introduction to the wound healing assay using live cell microscopy. Cell Adh Migr. 2014; 8: 440–451.
- Liang CC, Park AY, Guan JL. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007; 2: 329–333.
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP L complex inhibits RIPK3- dependent necrosis. Nature. 2011; 471: 363–367.
- Rubsam A, Parikh S, Fort PE. Role of Inflammation in Diabetic Retinopathy. Int J Mol Sci. 2018; 19: 942.
- Tisch N, Freire-Valls A, Yerbes R, Paredes I, Porta SL, Wang X, et al. Caspase-8 modulates physiological and pathological angiogenesis during retina development. J Clin Invest. 2019; 129: 5092–5107.
- Xuan YH, Huang BB, Tian HS, Chi LS, Duan YM, Wang X, et al. High- glucose inhibits human fibroblast cell migration in wound healing via repression of bFGF-regulating JNK phosphorylation. PloS one. 2014; 9: e108182
- Luthra S, Dong J, Gramajo AL, Chwa M, Kim DW, Neekhra A, Kuppermann BD, Kenney MC. 7-Ketocholesterol activates caspases-3/7, -8, and - 12 in human microvascular endothelial cells in vitro. Microvascular Research. 2008; 75: 343–350.









































































































































{kind=link}