Phenobarbital Toxicity Due to Poor Metabolism in a Neonate with Pyridoxine-Dependent Developmental and Epileptic Encephalopathy: A Case Report
- 1. Department of Pharmacy, Beijing Children’s Hospital, China
- 2. Neonatal Center, Beijing Children’s Hospital, China
- 3. Department of Stem Cell Transplantation, Beijing Children’s Hospital, China
- 4. Department of Neurology, Beijing Children’s Hospital, China
Abstract
Pyridoxine-dependent developmental and epileptic encephalopathy (PD-DEE) is a genetic metabolic disorder with seizures resistant to anti-seizure medications. We report a case of a male neonate with frequent focal motor seizures onset within the first day of life that were not controlled by conventional doses of phenobarbital. Moreover, he subsequently developed impaired consciousness and respiratory failure. His seizures stopped after an intravenous infusion of vitamin B6 at 17 days of age. A metabolic screening at 24 days of age revealed elevated pipecolic acid and he was started on oral vitamin B6. Genetic testing confirmed his diagnosis of PD-DEE caused by a pathogenic compound heterozygous variant in ALDH7A1. Meanwhile, a significantly elevated serum phenobarbital concentration was detected. After withdrawal of phenobarbital, his consciousness and respiration gradually returned to normal. Pharmacogenomic testing revealed that he carried several single nucleotide polymorphisms such as ABCB1 and SCN2A and was a poor metabolizer of phenobarbital, which explained the occurrence of toxicity. He was followed up to the age of 2 years and had no seizures or neurological sequelae with vitamin B6 supplementation. The case suggests that pharmacological doses of vitamin B6 are recommended in early-onset seizures, and genetic testing should be performed timely to identify the etiology and guide treatment decisions. Pharmacogenomic testing may be considered to guide individualized treatment when drug efficacy and adverse effects are significantly inconsistent with dosage and expectations.
CITATION
Zhang Y, Fang L, Nie H, Xu H, Weng J, Deng J (2024) Phenobarbital Toxicity Due to Poor Metabolism in a Neonate with Pyridoxine-Dependent Developmental and Epileptic Encephalopathy: A Case Report. Arch Paediatr Dev Pathol 7(1): 1028.
INTRODUCTION
Pyridoxine-dependent developmental and epileptic encephalopathy (PD-DEE, OMIM# 266100) is a genetic-metabolic disorder which was first reported in 1954 and the causative gene was identified as ALDH7A1 in 2006 [1]. ALDH7A1 encodes α-aminoadipic acid hemi-aldehyde dehydrogenase, the deficiency of which results in the accumulation of the intermediate products α-aminoadipic acid hemi-aldehyde, D1-piperidine-6-carboxylate and pipecolic acid, as well as the depletion of pyridoxine [2,3]. Most of the accumulated metabolites are neurotoxic, causing seizures and significant intellectual disability and developmental delay in 75% of patients [4]. Seizures in PD-DEE usually onset early and are characterized by resistance to anti-seizure medications (ASMs) but respond to pharmacological dose of pyridoxine [5]. Indeed, neonatal seizures may be the initial and predominant manifestation of inborn metabolic defects [6,7]. Therefore, if the neonatal seizures persist and resistant to conventional ASMs, congenital metabolic disorders should be considered [8]. In PD-DEE, early genetic diagnosis and timely pyridoxine supplementation are crucial to improve neurodevelopmental prognosis [9].
To date, phenobarbital (PB) remains the first line treatment for neonatal seizures [10]. It is metabolized mainly by the hepatic cytochrome enzyme CYP2C9 and to a lesser extent by CYP2C19 and CYP2E1 [11]. However, due to individual genetic polymorphisms, the encoded isoforms have different activities and respond to ASMs with great variability [7]. Pharmacogenomic testing is one of the outcomes of the high-throughput sequencing detection method, which can reflect the influence of oligonucleotide polymorphisms carried by individuals on pharmacokinetics and pharmacodynamics. Pharmacogenomic testing has recently been increasingly used in epilepsy, which may help to improve the safety and efficacy of ASM applications [7].
Here, we present a PD-DEE patient who developed toxicity due to reduced metabolic capacity for PB. This case suggests that pharmacogenomic testing may help the individualized treatment of epilepsy.
CASE PRESENTATION
The baby boy is the second child of a healthy nonconsanguineous Chinese Han couple with no relevant family history. At 38 weeks of gestation, he was found to have a fetal heart rate up to 160-180 beats per minute and was therefore delivered by emergency caesarean section. His birth weight was 2960 g. The Apgar score was 7 at 1 minute and 10 at both 5 and 10 minutes. Due to mild asphyxia, he was admitted to the neonatal intensive care unit at a local hospital and received nasal continuous positive airway pressure for assisted ventilation. He was fed formula milk through a nasogastric tube because he had difficulty sucking or swallowing. Fourteen hours after birth, he presented intermittent convulsions manifested by jerking of bilateral upper limbs and pedaling of bilateral lower limbs, which were focal onset motor seizures. A loading dose of PB 15 mg/kg was administered intramuscularly every 12 hours for 4 consecutive doses, followed by an oral maintenance dose of 5 mg/kg every 12 hours. Three days after birth, the boy continued to have recurrent convulsions with a frequency of more than 10 times per day, so he was sedated with intravenous midazolam (dose unknown), which resulted in a slight reduction in seizure frequency to 5-10 times per day.
The baby was transferred to our hospital 17 days of age and lethargy during the transfer. His convulsions stopped after 50 mg of vitamin B6 was administered intravenously in the emergency room, and he was subsequently admitted to the neonatal intensive care unit. On admission, he was weakly responsive to stimulation, with shallow and slow respiration, hypotonic extremities, and failure to elicit neonatal reflexes such as moro reflex. Laboratory tests revealed that the serum concentration of PB was higher than 80 μg/mL (reference 10–40 μg/mL). Therefore, PB was immediately withdrawn. Five and 10 days after the last dose, the re-examined PB concentration was 58.5 μg/mL and 22.9 μg/mL, respectively. As PB was gradually eliminated from his body, his consciousness improved, and he was no longer experiencing any seizures. Electroencephalography (EEG) performed at 18 days of age demonstrated delayed EEG maturation and multifocal epileptiform discharges during sleep [Figure 1].
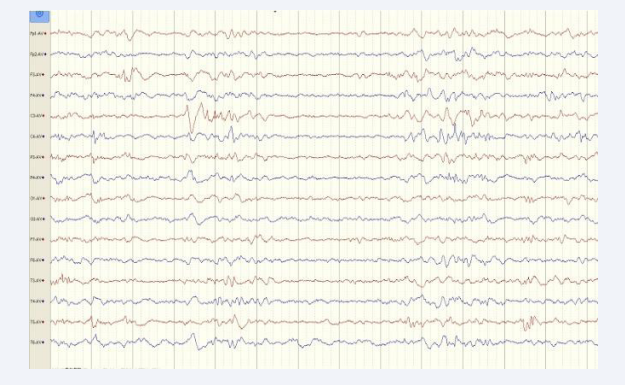
Figure 1: Electroencephalography performed at 18 days of age demonstrated multifocal epileptiform discharges during sleep.
Brain magnetic resonance imaging (MRI) performed at 19 days of age showed elevated T2 signal in the white matter, slightly higher T1 signal in the globus pallidus, and blurred long T1 signal in the posterior limbs of the internal capsule [Figure 2a].
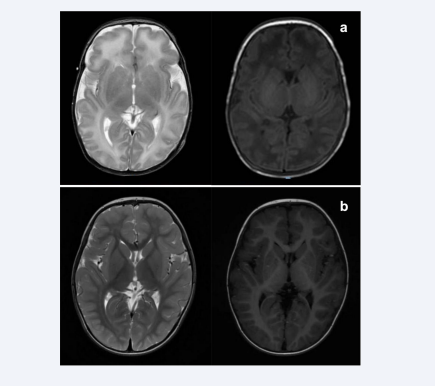
Figure 2: (a) Brain magnetic resonance imaging (MRI) performed at 19 days of age showed elevated T2 signal in the white matter (left), slightly higher T1 signal in the globus pallidus, and blurred long T1 signal in the posterior limbs of the internal capsule (right). (b) Re-examined brain MRI at the age of 2 years was normal.
At 24 days of age, metabolic screening showed elevated levels of pipecolic acid in both blood and urine, so he was started on oral vitamin B6 at a dose of 10 mg/kg.day.
Whole exome sequencing was performed and identified a compound heterozygous variant in ALDH7A1 of the patient, which was confirmed by Sanger sequencing. The variant c.1279C>G (p.Glu427Gln) was inherited from the asymptomatic father, which had a frequency of 0.000201 in the East Asian population of the gnomAD general population database and was recorded as pathogenic in the ClinVar database, meanwhile p.Glu427Gly and p.Glu427Asp at the same amino acid position have been reported in patients with PD-DEE [1,12]; c.1093+1C>T was inherited from the asymptomatic mother, which had a frequency of 0 in the East Asian population in gnomAD and was recorded as pathogenic in the ClinVar database, and it has been reported in patients PD-DEE [13,14]. According to the American College of Medical Genetics and Genomics [15], these two variants were both interpreted as pathogenic. Therefore, the infant was diagnosed with PD- DEE at 1.5 months of age. Meanwhile, pharmacogenomic testing was also performed, revealing that among the single nucleotide polymorphisms (SNPs) associated with PB metabolism, he carried ABCB1 rs1045642 AA, SCN2A rs17183814 GA, CYP2C19 rs6413438 CC and CYP2C19 rs4244285 GG.
To date, the boy has been followed up for 2 years. He is taking oral vitamin B6 at a dose of 10 mg/kg.day without any ASM. There were only 3 episodes of febrile convulsion at 1-year-old. His mental and motor development was mildly delayed, and his muscle tone was slightly low. However, after rehabilitation, he is now able to walk alone and speak several words. At the age of 2 years, his re-examined EEG and brain MRI were normal [Figure 2b].
DISCUSSION
With recent advances in pharmacogenomics, individualized and precision medicine are getting wider application in epilepsy treatment. This case reveals the importance of timely genetic testing in identifying etiology and predicting an individual’s drug response. Patients with PD-DEE are usually preterm and may present with signs of neonatal distress, irritability, and vomiting, sometimes accompanied by acidosis and low Apgar score, leading to a misdiagnosis of neonatal hypoxic-ischemic encephalopathy. After diagnosis, seizures control can be achieved with pharmacological dose of vitamin B6 in most patients. However, early onset PD-DEE often causes neurological damage that results in drug-resistant epilepsy if not correctly diagnosed and treated promptly [16]. Excessive fetal movements antenatally, mild neonatal asphyxia, frequent seizures onset within the first hours of life, and resistance to ASMs in this case were important clues to PD-DEE. Fortunately, his seizures were controlled by a single high dose of vitamin B6 given experimentally in the emergency room. Subsequently, after a timely metabolic screening and genetic test, the boy is correctly diagnosed and given vitamin B6 supplementation regularly. As a result, he is seizure-free and has no significant neurological sequelae. This case highlights the importance of metabolic investigation of elevated levels of α-aminoadipic semialdehyde and pipecolic acid in urine, plasma, or cerebrospinal fluid, as well as screening for ALDH7A1 variants in neonates and infants with unexplained and uncontrolled seizures with conventional treatment. High-dose vitamin B6 treatment should be tried in infants with early-onset and frequent seizures, especially if routine sedatives and ASMs are ineffective, even before a definitive diagnosis is made [17].
This patient presented with seizures in the neonatal period. Since PB is a first line treatment, he received conventional intramuscular loading doses and sequential oral maintenance doses of PB to control the seizures [10]. There was no overdose during treatment, but he had unexpected manifestations such as weakness and dyspnea with a significantly elevated PB serum concentration. Therefore, a pharmacogenomic test was performed and revealed that the boy carries ABCB1 rs1045642 AA, a SNP which leads to decreased intestinal expression of P-glycoproteins, resulting in elevated PB blood concentrations and drug susceptibility [18]. He also carries SCN2A rs17183814 GA which causes resistance to PB in east Asian population compared to GG type [19]; while CYP2C19 rs6413438 CC and CYP2C19 rs4244285 GG have not been reported as functional polymorphisms of cytochrome enzyme in Chinese Han population. Overall, pharmacogenomic test results suggest that he belongs to poor metabolism type of PB and has worse treatment response to PB, leading to an increased risk of adverse drug reactions at conventional doses [20]. The effects of multiple SNPs are independently superimposed, partially explaining the boy’s clinical manifestations. This case demonstrates that pharmacogenomic testing can be performed to aid individualize treatment in cases where ASMs are ineffective or significantly inconsistent with expectations, or when unexpected adverse effects occur [21]. PB is metabolized more rapidly in infants, with a half-life of about 67 hours, compared to 100 hours in adults [11], that is a hepatic enzyme inducer with interactions with other drugs. However, as one of the most used ASMs for neonatal convulsions, there is a lack of pharmacokinetic data for PB in the neonatal population, which may be limited by the difficulty in obtaining samples. Furthermore, pharmacogenomics still has major limitations at present, especially due to the lack of large- scale collaborative study, and the fact that effects of SNPs may vary between populations with different genetic backgrounds. We do not recommend pre-treatment pharmacogenomic testing for every patient, as this may delay treatment and cause unnecessary confusion and difficulty for drug choice. We believe that the selection of ASMs should be based on the principles of antiepileptic therapy, the experience of the physician and the individual circumstances of the patient, and that pharmacogenomic testing can be performed to help with dose adjustments or even therapy changes if unexpected outcomes and adverse effects occur.
CONCLUSION
Genomic testing and metabolic investigation can identify the diagnosis of congenital metabolic disorders, and treatment targeting the etiology may improve the effectiveness and safety of epilepsy therapy, thereby improving patients’ prognosis. In addition, pharmacogenomic testing can help guide individualized treatment when there is an inconsistency between drug efficacy or adverse effects with the dosage and blood concentration.
DECLARATIONS
Author contribution statement
All authors listed have significantly contributed to the investigation, development and writing of this article.
Funding statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Ethics statement
Ethical approval for this study was obtained from the Ethics Committee of Beijing Children’s Hospital. Verbal informed consent was obtained from the guardians for the publication of all images, clinical data, and other data contained in the manuscript.
REFERENCES
- Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006; 12: 307-309.
- Coughlin CR 2nd, Swanson MA, Spector E, Meeks NJL, Kronquist KE, Aslamy M, et al. The genotypic spectrum of ALDH7A1 mutations resulting in pyridoxine dependent epilepsy: A common epileptic encephalopathy. J Inherit Metab Dis. 2019; 42: 353-361.
- Bok LA, Halbertsma FJ, Houterman S, Wevers RA, Vreeswijk C, Jakobs C, et al. Long-term outcome in pyridoxine-dependent epilepsy. Dev Med Child Neurol. 2012; 54: 849-854.
- Kaminiów K, Paj?k M, Paj?k R, Paprocka J. Pyridoxine-Dependent Epilepsy and Antiquitin Deficiency Resulting in Neonatal-Onset Refractory Seizures. Brain Sci. 2021;12: 65.
- Haidar Z, Jalkh N, Corbani S, Fawaz A, Chouery E, Megarbane A. Atypical pyridoxine dependent epilepsy resulting from a new homozygous missense mutation, in ALDH7A1. Seizure. 2018;57:32- 33.
- Sharma S, Prasad AN. Inborn Errors of Metabolism and Epilepsy: Current Understanding, Diagnosis, and Treatment Approaches. Int J Mol Sci. 2017; 18: 1384.
- Balestrini S, Sisodiya SM. Pharmacogenomics in epilepsy. Neurosci Lett. 2018; 667: 27-39.
- Rahman S, Footitt EJ, Varadkar S, Clayton PT. Inborn errors of metabolism causing epilepsy. Dev Med Child Neurol. 2013; 55: 23-36.
- Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63: 1349-1397.
- Samanta D. Recent Advances in the Diagnosis and Treatment of Neonatal Seizures. Neuropediatrics. 2021; 52: 73-83.
- Pacifici GM. Clinical Pharmacology of Phenobarbital in Neonates: Effects, Metabolism and Pharmacokinetics. Curr Pediatr Rev. 2016; 12: 48-54.
- Coulter-Mackie MB, Li A, Lian Q, Struys E, Stockler S, Waters PJ. Overexpression of human antiquitin in E. coli: enzymatic characterization of twelve ALDH7A1 missense mutations associated with pyridoxine-dependent epilepsy. Mol genet metab. 2012; 106: 478-481.
- Butler KM, Silva Cd, Alexander JJ, Hegde M, Escayg A. Diagnostic Yield From 339 Epilepsy Patients Screened on a Clinical Gene Panel. Pediatr neurol. 2017; 77: 61-66.
- Scharer G, Brocker C, Vasiliou V, Creadon-Swindell G, Gallagher RC, Spector E, et al. The genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy due to mutations in ALDH7A1. J inherit metab dis. 2010;33: 571-581.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine: official journal of the American College of Medical Genetics. 2015; 17: 405-424.
- Saracino MA, Tallarico K, Raggi MA. Liquid chromatographic analysis of oxcarbazepine and its metabolites in plasma and saliva after a novel microextraction by packed sorbent procedure. Anal chim acta. 2010; 661: 222-228.
- Cirillo M, Venkatesan C, Millichap JJ, Stack CV, Nordli DR, Jr. Case Report: Intravenous and Oral Pyridoxine Trial for Diagnosis of Pyridoxine-Dependent Epilepsy. Pediatrics. 2015; 136: 257-261.
- Lakhan R, Misra UK, Kalita J, Pradhan S, Gogtay NJ, Singh MK, et al. No association of ABCB1 polymorphisms with drug-refractory epilepsy in a north Indian population. Epilepsy Behav. 2009; 14: 78-82.
- Lakhan R, Kumari R, Misra UK, Kalita J, Pradhan S, Mittal B. Differential role of sodium channels SCN1A and SCN2A gene polymorphisms with epilepsy and multiple drug resistance in the north Indian population. Br J Clin Pharmacol. 2009; 68: 214-220.
- Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017; 140: 1316-1336.
- Patsalos PN, Spencer EP, Berry DJ. Therapeutic Drug Monitoring of Antiepileptic Drugs in Epilepsy: A 2018 Update. Ther Drug Monit. 2018; 40: 526-548.









































































































































{kind=link}