Whole-Exome Sequencing in a Pediatric Patient with Relapsed or Refractory Burkitt Leukemia Resistant to Conventional Chemotherapy
- 1. Department of Pediatrics, Gunma University Graduate School of Medicine, Japan
- 2. Department of Hematology/Oncology, Gunma Children’s Medical Center, Japan
- 3. Clinical Research Center, National Hospital Organization Nagoya Medical Center, Japan
- 4. Department of Pathology and Tumor Biology, Kyoto University, Japan
- 5. Laboratory of DNA Information, The University of Tokyo, Japan
- 6. Department of Pediatrics, Takasaki General Medical Center, Japan
- 7. Department of Pediatrics Hematology and Oncology Research, National Center for Child Health and Development, Japan
- 8. Director General, Japanese Red Cross Gunma Blood Center, Japan
- 0. These authors contributed equally to this work
Abstract
Burkitt lymphoma/leukemia (BL) is one of the most frequent B-cell lymphoma types in children. The cure rate for pediatric BL has improved in the last 30 years; long-term event-free survival of patients has reached approximately 90%. However, novel treatment approaches are needed for a group of high-risk patients. Here, we report the case of a pediatric patient with relapsed/refractory BL who died despite undergoing multimodal treatment including clofarabine. To determine the reason for this resistance, we performed whole-exome sequencing using primary, relapsed, and complete remission samples, and identified 34 gene mutations including TP53, ID3, ARID1A, CCND3, DDX3X, MYC and CHD6. Relapsed BL seemed to evolve from one of the subclones observed at the initial phase and was accompanied by many additional mutations including the chromatin remodeling mutation that was absent or existed at a lower allele frequency in the diagnostic samples, indicating a multistep process of BL recurrence.
Citation
Kawashima J, Okuno H, Shiba N, Yoshida K, Shiraishi Y, et al. (2017) Whole-Exome Sequencing in a Pediatric Patient with Relapsed/Refractory Burkitt Leukemia Resistant to Conventional Chemotherapy. Arch Paediatr Dev Pathol 1(1): 1001.
Keywords
• Burkitt lymphoma/leukemia
• Clofarabine
• Whole-exome sequencing
• ID3
• MYC
ABBREVIATIONS
BL: Burkitt lymphoma/leukemia; CLO: Clofarabine; SCT: Stem Cell Transplantation; NHL: Non-Hodgkin Lymphoma; CR: Complete Remission; WES: Whole-exome Sequencing; WBC: White Blood Cell Count; CNS: Central Nerve System; R-ICE: Rituximab, Ifosfamide, Carboplatin, and VP-16; CY: Cyclophosphamide; BMT: Bone Marrow Transplantation; B-NHL: B-cell Non-Hodgkin Lymphoma
INTRODUCTION
Burkitt lymphoma/leukemia (BL) is one of the most frequent types of B-cell lymphoma in children. The EpsteinBarr virus (EBV) and MYC translocation have been considered to be important players in BL pathogenesis, neither is sufficient to explain the behavior of the tumor [1]. t(8;14) has also been detected in blood cells of healthy individuals, reinforcing the idea that it alone is not sufficient to drive tumor development [2]. The high proliferation of BL cells could be related to dysregulation of the cell cycle induced by the MYC protein, but MYC also activates apoptosis; therefore, BL cells must have mechanisms to overcome this tumor-protective effect [1]. The cure rate for pediatric BL has improved in the last 30 years, and long-term event-free survival of patients has reached approximately 90% [3-6]. However, there remains a group of high-risk patients for whom novel treatment approaches are needed [7]. The efficacy of rituximab combination chemotherapy as a first-line or salvage therapy for various types of CD20-positive non-Hodgkin’s lymphoma, including BL, has been described in recent reports [8,9].
Recently, we experienced the case of a relapse/refractory BL patient who was refractory to chemotherapy and died despite undergoing multimodal treatment including rituximab, clofarabine (CLO), and stem cell transplantation (SCT) from HLA 2-locus mismatched related donor. Currently, no standard therapy exists for patients with relapsed and/or refractory nonHodgkin lymphoma (NHL) who are ineligible for transplantation or who have failed to obtain complete remission (CR) by SCT. To obtain a better understanding of the correlation between gene mutations and clinical course and explore the molecular targets, trio tumor-normal-relapsed DNA specimens were analyzed using whole-exome sequencing (WES).
CASE PRESENTATION
Clinical course
A 9-year-old Japanese boy was admitted to our hospital with complaints of fever, nausea, vomiting, weight loss, and stomachache. He was pale and had petechiae, and a mass around the ileocecum. The results of laboratory testing were as follows: White blood cell (WBC) count, 41.7 × 109 /L; hemoglobin, 12.7 g/dL; Platelet count, 58.0 × 109 /L; LDH level, 11,863 U/L; and uric acid level, 13.6mg/dL. Bone marrow smear preparation revealed that more than 90% of the cells were lymphoid blasts with myeloperoxidase negative (Figure 1). Surface marker analysis showed that the leukemic blasts in the bone marrow were positive for CD5 (0.6%), CD10 (34.7%), CD19 (93.3%), CD20 (89.2%), CD22 (37.1%), κ-ch. (1.6%), λ-ch. (96.3%), CD13 (3.6%), CD38 (97.8%), CD56 (0.7%), FMC-7 (76.9%), and HLADR (97.2%) antigens. Chromosomal analysis of bone marrow cells revealed 46, XY, +1, der(1;22)(q10;q10), t(8;14)(q24;q32). EBV was not detected in the peripheral blood or bone marrow. Leukemic cells invaded the central nerve system (CNS) (25/ul). The patient was diagnosed with CNS-positive stage IV BL, i.e., Group 4 according to the Japan Pediatric Leukemia/Lymphoma Study Group B-NHL03 clinical study. The treatment schedule was reported previously [10,11]. We safely used rasburicase (0.2 mg/ kg) and thrombomodulin (380 U/kg/day) therapy to prevent tumor lysis syndrome progression. Following the initial 4A1 treatment, the patient achieved CR. Although he could maintain CR successfully for 4 months, BL suddenly relapsed after the fourth consolidation therapy (Figure 1).
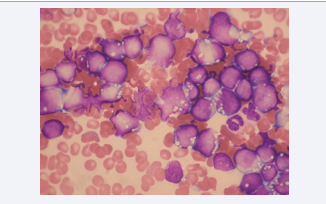
Figure 1 The light microscopic image of the Burkitt leukemia cells. Wright’s stain of bone marrow aspiration revealed almost all L3 blast cells.
The results of the blood test at relapse were as follows: WBC count, 18.1 × 109 /L, LDH level of 7,029 U/L, and uric acid level of 7.2 mg/dL. We used rasburicase (0.2 mg/kg) again to prevent tumor lysis syndrome without any anaphylactic reaction, as well as attempted some other regimens, including rituximab, ifosfamide, carboplatin, and VP-16 (R-ICE); however, another CR could not be achieved. Furthermore, the leukemic cells had down-regulated their CD20 expression after six courses of rituximab treatment. To overcome this situation, we tried to use CLO with cyclophosphamide (CY) and VP-16 (CLO 40 mg/m2 , CY 440 mg/m2 , and VP-16 100 mg/m2 for 5 days) [12]. We safely administered these drugs for 5 days as planned initially and the WBC count was maintained at 0/L for 2 weeks without a blast crisis. No side effects were observed except grade 4 blood toxicity. We continuously performed allo-bone marrow transplantation (BMT) donated from an HLA 2-locusmismatched related donor. The conditioning regimen consisted of total body irradiation and melphalan. BMT with 3.6 × 106 CD34 cells/kg was performed. We could not confirm the engraftment, and a blast crisis occurred concomitantly. The patient died on day 51 after allo-BMT (Figure 2).
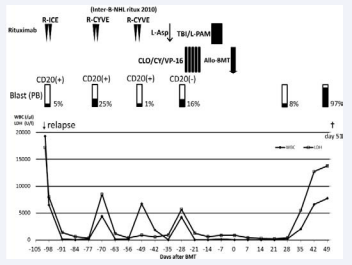
Figure 2 Clinical course of the patient after relapsed. TBI indicates total body irradiation; BMT, bone marrow transplantation; WBC, white blood cell count; PB, peripheral blood; BD, bortezomib and dexamethasone; ICE, ifosfamide, carboplatin, and VP-16; CYVE, Ara-C x2 and VP-16; L-asp, L-asparaginase; L-PAM, melphalan; CLO, clofarabine.
Whole-exome sequencing analysis
To reveal the clonal origin and major mutational events in relapsed pediatric BL, we performed WES in this patient. Tumour DNA was extracted from the patient’s bone marrow mononuclear cells at diagnosis and at the relapse phase, and germline control DNA was obtained from the patient’s bone marrow mononuclear cells at CR. Whole-exome capture was accomplished based on liquid phase hybridization of sonicated genomic DNA with a mean length of 150–200 base points (bp) to the bait cRNA library synthesized on magnetic beads (Sure Select Human All Exon 50Mb or V5 kit®, Agilent Technology, CA, USA), according to the manufacture’s protocol. Captured targets were subjected to massive sequencing using Illumina HiSeq 2000 with the paired-end 100 bp read option, according to the manufacturer’s instructions. WES of three specimens obtained at diagnosis, relapse, and in CR phases were analyzed with a mean coverage of more than ×100, and 95% of the targeted sequences were analyzed at an average depth of more than ×20. Data processing and variant calling were performed as previously described [13]. Candidate somatic mutations were detected using our inhouse pipeline Empirical Bayesian mutation Calling (EBCall; see URLs; http://genomon.hgc.jp/exome/) [14]. All candidates were validated by Sanger sequencing. We identified 34 gene alterations including TP53, ID3, ARID1A, DDX3X, MYC, and CHD6 (Figure 3).
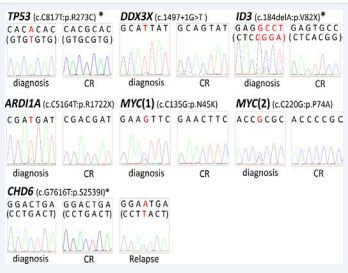
Figure 3 Chromatogram of sequencing with fluorescent-dye chemistry for unfractionated genomic DNA from a Burkitt leukemia patient. *In ID3, TP53, and CHD6, Sanger sequence analysis was performed using their reverse primers. Nucleotide in the parenthesis in each gene shows the coding strand sequence. CR indicates complete remission.
Of the 34 mutations identified, 10 were specific to relapse (LRRC36, FAM155B, PHF14, KLF17, AKAP9, CHD6, ZIC1, RHBDF1, HIST1H2BJ, and EEA1), whereas two mutations (LIM2 and RFX7) were specific to the time of diagnosis (Figure 4 and Table 1). Relapsed BL evolved from one of the subclones observed at the initial phase and was accompanied by many additional mutations.
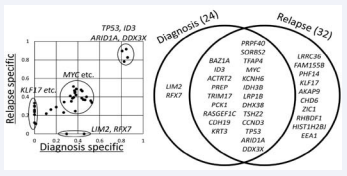
Figure 4 Whole-exome sequencing in a pediatric patient with Burkitt leukemia. Variant allele frequencies of validated gene mutations were shown in a diagonal plot. Thirty-four mutations identified by whole-exome sequencing were shown in Venn diagram.
|
Table 1: Validated 34 somatic mutations in a pediatric patient with Burkitt leukemia. |
|||||
|
Gene |
Function |
Gene number |
Change |
Result |
Chromosome |
|
LIM2 |
missense |
NM_030657 |
c.C293A:p.A98E |
diagnostic_specific |
19 |
|
RFX7 |
missense |
NM_022841 |
c.G816T:p.M272I |
diagnostic_specific |
15 |
|
LRRC36 |
missense |
NM_001161575 |
c.C1231A:p.L411I |
relapse_specific |
16 |
|
FAM155B |
missense |
NM_015686 |
c.C940A:p.Q314K |
relapse_specific |
X |
|
PHF14 |
missense |
NM_014660 |
c.G2059A:p.G687S |
relapse_specific |
7 |
|
KLF17 |
missense |
NM_173484 |
c.A1123G:p.N375D |
relapse_specific |
1 |
|
AKAP9 |
missense |
NM_005751 |
c.A4373T:p.Q1458L |
relapse_specific |
7 |
|
CHD6 |
missense |
NM_032221 |
c.G7616T:p.S2539I |
relapse_specific |
20 |
|
ZIC1 |
missense |
NM_003412 |
c.A533G:p.Y178C |
relapse_specific |
3 |
|
RHBDF1 |
missense |
NM_022450 |
c.G908A:p.G303D |
relapse_specific |
16 |
|
HIST1H2BJ |
missense |
NM_021058 |
c.C120G:p.I40M |
relapse_specific |
6 |
|
EEA1 |
nonsense |
NM_003566 |
c.G1396T:p.E466X |
relapse_specific |
12 |
|
BAZ1A |
frameshift insertion |
NM_013448 |
c.262_263insT:p.P88fs |
14 |
|
|
ID3 |
missense |
NM_002167 |
c.184delA:p.V82X |
1 |
|
|
ACTRT2 |
missense |
NM_080431 |
c.G159C:p.Q53H |
1 |
|
|
PREP |
missense |
NM_002726 |
c.G1522A:p.G508S |
6 |
|
|
TRIM17 |
missense |
NM_001024940 |
c.G1335C:p.Q445H |
1 |
|
|
PCK1 |
missense |
NM_002591 |
c.A989G:p.N330S |
20 |
|
|
RASGEF1C |
missense |
NM_175062 |
c.G1144A:p.A382T |
5 |
|
|
CDH19 |
missense |
NM_021153 |
c.A656C:p.E219A |
18 |
|
|
KRT3 |
missense |
NM_057088 |
c.G1126A:p.A376T |
12 |
|
|
PRPF40A |
nonsense |
NM_017892 |
c.C1735T:p.R579X |
2 |
|
|
SORBS2 |
missense |
NM_001145674 |
c.A2843G:p.E948G |
4 |
|
|
TFAP4 |
missense |
NM_003223 |
c.C172T:p.R58W |
16 |
|
|
MYC |
missense |
NM_002467 |
c.C135G:p.N45K |
8 |
|
|
MYC |
missense |
NM_002467 |
c.C220G:p.P74A
|
|
8 |
|
KCNH6 |
missense |
NM_030779 |
c.C2681T:p.P894L |
17 |
|
|
IDH3B |
nonsense |
NM_174856 |
c.499delA:p.I167X |
20 |
|
|
LRP1B |
missense |
NM_018557 |
c.A10571G:p.N3524S |
2 |
|
|
DHX38 |
missense |
NM_014003 |
c.C1099T:p.R367W |
16 |
|
|
TSHZ2 |
missense |
NM_001193421 |
c.G725A:p.R242H |
20 |
|
|
CCND3 |
nonsense |
NM_001136125 |
c.C622T:p.Q208X |
6 |
|
|
TP53 |
missense |
NM_001126115 |
c.C817T:p.R273C |
17 |
|
|
ARID1A |
nonsense |
NM_006015 |
c.C5164T:p.R1722X |
1 |
|
|
DDX3X |
splicing |
NM_001193416 |
c.1497+1G>T |
? |
X |
DISCUSSION
We experienced the case of pediatric patient with relapsed/ refractory BL who finally died despite undergoing multimodal treatment including CLO. In the present study, a total of 34 somatic mutations were identified, where the number of nonsynonymous mutations was higher in the relapse phase than at the time of diagnosis (24 vs. 32) (Figure 4 and Table 1). Among them, thus far, ID3, CCND3, DDX3X, and MYC were identified as the top four recurrent gene mutations found in four prototypical BL with IGMYC translocation [7]. Love et al. also reported almost similar genetic landscape of mutations in BL using next-generation sequencing [15]. In addition to these mutations, mutations of TP53 and ARID1A, which are known as suppressor genes, were also identified [7,15]. Abate et al. detected lower frequencies of mutations in MYC, ID3, TCF3, and TP53 and a higher frequency of mutations in ARID1A in endemic BL samples [16]. This result indicates dual mechanisms of transformation, i.e., mutation versus virus-driven mechanisms in sporadic and endemic BL [16]. ID3 mutations occur in 34%–68% of BL and is identified at a greater frequency in adult samples [7]. ID3 acts a negative transcriptional regulator by sequestering transcription factors with basic helix–loop–helix motifs. Mutated ID3 attenuates this regulatory interaction [17,18]. ID3 and its interaction partner TCF3 are involved in controlling cell cycle progression and survival pathways through tonic B-cell signaling [19,20]. CCND3 and ID3 double-hit mutations, as well as 18q21 cytogenetically normal-loss of heterogeneity, have been reported to be associated with poorer outcome [20].
Concerning allele frequencies, intratumoral gene mutations were observed to be heterogeneous, suggesting multiple clonal evolution events during the development of BL (Figure 4). Variant allele frequency of mutated TP53, ID3, ARID1A, and DDX3X alleles were higher than that of MYC, IDH3B, PRPF40A, and LIM2. These results suggest that the mutation of TP53, ID3, ARID1A, and DDX3X occurred at an early stage in leukemic cells, whereas the mutation of MYC, IDH3B, PRPF40A, and LIM2 occurred as a secondary event. Cells from the founding clone can acquire additional cooperating mutations, yielding subclones that can contribute to disease progression. Notably, we identified mutation of CHD6 only at the relapsed phase, which is one of the chromatin remodeling genes, as well as ARID1A [21]. This results suggested that aberration of chromatin regulation might be associated with disease progression. Relapse may involve dynamic clonal changes following combination chemotherapy and is accompanied by many additional mutations including the chromatin remodeling mutation that were absent or existed at a lower allele frequency in the diagnostic samples, indicating a multistep process of BL recurrence.
Rituximab is a chimeric monoclonal antibody that recognizes the CD20 antigen and is used to treat B-cell NHL (B-NHL) including BL. Although most of the cases benefit from this agent, some cases become resistant to this agent. Few studies have been published which evaluate B-NHL treated with rituximab, and showed that CD20 expression decreased or was lost in many cases of B-NHL persisting after rituximab therapy [22]. In fact, CD20 expression was lost after combination chemotherapy including rituximab in our patient, leading to resistance to conventional chemotherapy. In such severe circumstances, we could safely and effectively use CLO to treat heavily pretreated pediatric patients with relapsed/refractory BL harboring a lot of oncogenic mutations. CLO is tolerated well without significant adverse events, except for grade 4 reversible myelosuppression. We observed that CLO administration provided a favorable response in patients with NHL who have relapsed after receiving multiple therapies and who were refractory to rituximab. Combination therapy is the current standard of front-line treatment for patients with NHL. Therefore, the present case suggests that various CLObased combination regimens, specifically those with monoclonal antibodies, are worth exploring in refractory patients who are resistant to multiple conventional chemotherapy and rituximab. We consider that this agent effectively reduces leukemic cells when used with cyclophosphamide and VP-16.
In conclusion, from the viewpoint of molecular analysis, WES revealed 34 gene mutations including TP53, ID3, ARID1A, CCND3, DDX3X, CHD6 and MYC, and relapsed BL seemed to evolve from one of the subclones observed at the initial phase and was accompanied by many additional mutations including the chromatin remodelling mutation that were absent or existed at a lower allele frequency in the diagnostic samples, indicating a multistep process of BL recurrence.
ACKNOWLEDGMENTS
The authors would like to thank Yuki Hoshino for her excellent assistance and Enago (www.enago.jp) for the English language review.







































































































































{kind=link}