Assembly of Antibody-Drug Conjugates as Potent Immunotherapy
- 1. Chengdu Institute of Biology, Chinese Academy of Sciences, China
- 2. Center for Growth, Metabolism and Aging, College of Life Sciences, Sichuan University, China
Abstract
Arming antibodies with potent cytotoxic drugs greatly enhances the activity of antibodies, while reduces the systemic toxicity of cytotoxic drugs. Resent approvals of antibody-drug conjugates (ADCs) by FDA and the good performance of ADCs in clinical trials indicate promising potential of ADCs as immunotherapeutics. Safety and efficacy of ADCs are greatly influenced by their pharmacokinetics, in which the linkage between the antibody and the drug plays a critical role. This review discusses the progress in linkage optimization with an emphasis on the development of new ADCs with better safety and efficacy. It includes the exploration of drug release mechanism in the drug side of the linker, the superiority of site-specific ADCs over conventional ADCs, and the application of site-specific protein modification technology to generate site-specific ADCs. Related advances in molecular biology and chemical biology that benefit this field are also discussed.
Keywords
Antibody-drug conjugate; Linker; Site-specific antibody-drug conjugate; Better safety and efficacy.
Citation
Chen S, Cao Y (2014) Assembly of Antibody-Drug Conjugates as Potent Immunotherapy. JSM Cell Dev Biol 2(1): 1006.
ABBREVIATIONS
ADC: Antibody-Drug Conjugate
INTRODUCTION
Upon binding to tumor cells, monoclonal antibodies (Mabs) can activate NK cells and complement pathway to induce tumor cell lysis, and modulate tumor-related signaling by blocking ligand binding [1]. However, their curative potential is limited and considerable efforts have been devoted to conjugating antibodies with potent small molecular cytotoxic drugs [2]. Antibody-drug conjugate (ADC) not only enhances the activity of antibodies, but also improves the selectivity of cytotoxic drugs and reduces their systemic toxicity, making it a promising immunotherapy strategy [3,4]. Currently, there are two ADCs, brentuximab vedotin and trastuzumab emtansine approved by FDA and over 20 ADCs in clinical development [5,6] (Table 1).
Table 1: Approved ADCs and ADCs in Clinical Development.
| ADC | Target | Drug | Linker | Indication | Sponsor |
| Approved | |||||
| Trastuzumab emtansine | HER2 | Maytansine DM1 | Non-cleavable Linker | Breast Cancer | Genentech/Roche |
| Brentuximab vedotin | CD30 | Auristatin MMAE | Valine-citrulline | HL/ALCL | Seattle Genetics |
| in Clinical Development | |||||
| Inotuzumab ozogamicin | CD22 | Calicheamicin | Hydrazone | NHL | Pfizer |
| Gemtuzumab ozogamicin | CD33 | Calicheamicin | Hydrazone | Relapsed AML | Pfizer |
| SAR3419 | CD19 | Maytansine DM4 | Disulfide Linker SPDB | NHL | Sanofi |
| SAR566658 | CA6 | Maytansine DM4 | Disulfide Linker SPDB | Solid Tumors | Sanofi |
| BT062 | CD138 | Maytansine DM4 | Disulfide Linker SPDB | MM | Biotest |
| AMG 172 | CD27L | Maytansine DM1 | Non-cleavable Linker | Kidney Cancer | Amgen |
| AMG 595 | EGFRvIII | Maytansine DM1 | Non-cleavable Linker | Recurrent Gliomas | Amgen |
| RG-7593 | CD22 | Auristatin MMAE | Valine-citrulline | NHL | Genentech/Roche |
| RG-7596 | CD79b | Auristatin MMAE | Valine-citrulline | NHL | Genentech/Roche |
| SGN-75 | CD70 | Auristatin MMAF | Non-cleavable Linker | RCC, NHL | Seattle Genetics |
| SGN-CD19A | CD19 | Auristatin MMAF | Non-cleavable Linker | Leukemia, Lymphoma | Seattle Genetics |
| SGN-CD33A | CD33 | PBD dimer | Valine-alanine | AML | Seattle Genetics |
| SGN-LIV1A | LIV-1 | Auristatin MMAE | Peptide Lnker | Breast Cancer | Seattle Genetics |
| Milatuzumab doxorubicin | CD74 | Doxorubicin | Hydrazone | MM | Immunomedics |
| IMMU-130 | CEACAM5 | SN38 | Phenylalanine-lysine | CRC | Immunomedics |
| IMMU-132 | TROP2 | SN-38 pH | Sensitive Linker | Epithelial Cancers | Immunomedics |
| Lorvotuzumab mertansine | CD56 | Maytansine DM1 | Disulfide Linker SPP | Solid tumors, MM | Immunogen |
| IMGN289 | EGFR | Maytansine DM1 | Non-cleavable Linker | Solid Tumors | Immunogen |
| IMGN853 | FOLR1 | Maytansine DM4 | Disulfide Linker sulfo-SPDB | Solid Tumors | Immunogen |
| Glembatumomab vedotin | GPNMB | Auristatin MMAE | Valine-citrulline | Breast Cancer, Melanoma | Celldex Therapeutics |
| PSMA ADC | PSMA | Auristatin MMAE | Valine-citrulline | Prostate Cancer | Progenics |
| AGS-5ME | SLC44A4 | Auristatin MMAE | Valine-citrulline | Pancreatic, Prostate Cancer | Astellas |
| ASG-22ME | Nectin-4 | Auristatin MMAE | Valine-citrulline | Solid Tumors | Astellas |
| AGS-16C3F | ENPP3 | Auristatin MMAF | Non-cleavable Linker | RCC | Astellas |
| BAY 79-4620 | CA-IX | Auristatin MMAE | Valine-citrulline | Solid Tumors | Bayer |
| BAY 94-9343 | Mesothelin | Maytansine DM4 | Disulfide Linker SPDB | Solid Tumors | Bayer |
Abbreviations: ADC: antibody-drug conjugate; SN-38: the active metabolite of irinotecan, a potent topoisomerase I inhibitor; PBD dimer: potent DNA cross-linking pyrrolobenzodiazepine dimmer; HL: Hodgkin’s Lymphoma; ALCL: Anaplastic Large Cell Lymphoma; NHL: Non-Hodgkin’s Lumphoma; AML: Acute Myelogenous Leukemia; MM: Multiple Myeloma; RCC: Renal Cell Carcinoma; CRC: Colorectal Carcinoma.
An ADC is composed of an antibody, the small molecular cytotoxic drug and a linker. The cytotoxic drugs are internalized into targeted tumor cells and trafficked to the lysosome with antibodies by antibody mediated receptor endocytosis and released thereafter [7]. In order to achieve significant cytotoxicity, selection of cytotoxic drugs has been shifted from traditional chemotherapeutics to highly potent cytotoxic drugs, such as calicheamicins, auristatins and maytansinoids, which are too toxic to use independently [8]. The use of highly potent drugs presents big challenges on the design of linkers which must ensure toxic drugs are only released at the tumor site [9]. In the past decade, significant progress has been made including the investigation of new antibody targets [10] and antibody formats [11], the selection of potent cytotoxic drugs [2], the optimization of the linkage between antibodies and drugs by exploring drug release mechanisms and developing the conjugation technology as well as site-specific antibody modification technology [12].
In this review, we focus on the optimization of the linkage between the antibody and the drug, with an emphasis on the development of new promising ADCs with better safety and efficacy, including the exploration of drug release mechanism in the drug side of the linker, the superiority of site-specific ADCs over conventional ADCs, and the application of site-specific protein modification technology on ADCs.
Linkage between the antibody and the drug
The small molecular drug is attached to the antibody by a bifunctional linker, which contains distinct modules (Figure 1).
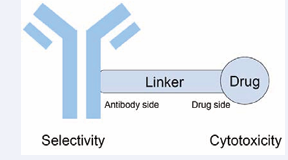
Figure 1: Illustration of antibody drug conjugates.
The module that links to the drug (the drug side) controls the selective release of a drug at the tumor site. The key of its design is to take advantage of differences between the blood and the intracellular environment. The module that links to the antibody (the antibody side) involves modification of antibodies and will be discussed in following sections.
Up to now, there have been several strategies developed in designing the drug side of linkers. 1) Lysosomes (pH 4.5–5.0) and endosomes (pH 5.5–6.2) provide low pH environments whereas the systemic circulation (pH 7.4–7.5) does not. Acid-labile hydrazone linkers are employed to selectively release drugs inside cells due to the pH difference [13]. 2) The intracellular cytosol, which contains large numbers of reducing molecules such as glutathione, is a significantly reducing environment compared to the systemic circulation. Based on this property, disulfide linkers are designed, with varying degrees of steric hindrance to further improve their stability in the circulation [14]. 3) Using peptide linkers cleavable by the intracellular lysosomal proteases is another selective drug release mechanism [15]. Lysosomal proteases, such as cathepsin B, are highly expressed in lysosomes and are only active in low pH environments. The dipeptide sequence of valine-citrulline is an optimized choice for peptide linkers [16], with a para-aminobenzyl (PAB) group which is used as a self-immolative spacer to help drug release. 4) Other than above cleavable linkers, an alternative strategy is to use non-cleavable linkers for drugs which could keep their potency after modification of the linker. Non-cleavable linkers do not contain any drug release triggers. Drugs are released when the internalized antibodies are degraded in lysosomes, leaving the drug still connected with the linker [17]. Peptide linkers and non-cleavable linkers are regard to be the most successful linker design in the drug side, for many in vivo experiments showed that linkers using these two drug release mechanism had higher stability in the systemic circulation and a much longer half life [2,18].
Site-specific antibody-drug conjugates
Small molecular drugs are generally conjugated to the sidechain amines of lysine residues or the thiol group of cysteines (generated by reducing interchain disulfide bonds) on the antibody. The drug conjugation process yields heterogenous products, a mixture of ADCs with various conjugation sites and different molar ratios of drug per antibody, because there are abundant lysines and disulfide bonds in antibodies. It has been shown that even conjugating through the less abundant cysteine residues by partial reduction of interchain-disulfide bonds, the products were highly heterogenous, potentially containing over 100 distinct species [19-20], not to mention conjugating through the lysine residue. Different species in the ADC products vary in pharmacokinetic, efficacy and safety, bringing great challenges for quality control of ADCs. Besides, heavily loaded conjugates were discovered to be rapidly cleared from the circulation in vivo [21]. Thus site-specific conjugation is a better strategy because it generates ADCs with high homogeneity for both conjugation site and stoichiometry of drugs.
It is an efficient site-specific protein modification strategy to incorporate a cysteine residue at a chosen site for labeling its reactive thiol by using thiol-maleimide chemistry under a mild reaction condition [22]. Based on this strategy, Junutula et al developed site-specific ADCs, called THIOMABs, by using linkers containing maleimide on the antibody site to conjugate with incorporated cysteines [19] (Figure 2). Suitable conjugation sites were screened by a phage display-based method [23]. Mild reduction and reoxidation were carried out to keep native interchain disulfide bonds undisrupted and thus restricted the conjugation to the incorporated cysteines (Figure 2).
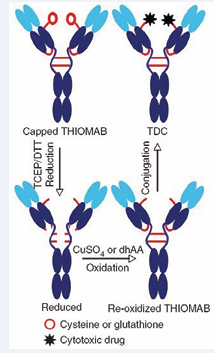
Figure 2: Illustration of THIOMAB, a site-specific ADCs technology developed by Junutula et al [19]. This figure is adapted from [19]. The ADC was generated by site-specifically incorporating a cysteine residue, with which drugs are conjugated through thiol-reactive linkers. Mild reduction and reoxidation were carried out to keep native interchain disulfide bonds undisrupted.
Functional assays of the conjugated antibodies demonstrated that the cysteine substitution and the conjugation retained the antibody’s properties, including the affinity, specificity, and internalization rate. In the example of an anti-MUC16 THIOMAB, in which two cysteine residues were incorporated and two moles of monomethyl auristatin E (MMAE) were conjugated per antibody, analysis of conjugation products exhibited near-homogenous composition. This site-specific ADC displayed comparable efficacy to its conventional ADC counterpart in mouse xenograft models. In the safety study using rats and cynomolgus monkeys, this site-specific ADC was better tolerated than the conventional ADC [19], indicting improved safety.
Although the connection of the thiol group in the antibody and the thiol-reactive linkers was always supposed to be stable in physical conditions, a research on selection of conjugation sites by Shen et al. [24] revealed that the stability of this connection may not be so stable as supposed, as it was interfered by reactive thiols in the systemic circulation and was modulated by the structural and chemical environment surrounding the conjugation site. An anti-Her2 antibody trastuzumab conjugated with monomethyl auristatin E (MMAE) using thiol-maleimide chemistry with different conjugation sites varying in solvent accessibility and local charge were studied by Shen et al. They found that thiol-reactive linkers with cytotoxic drugs could transfer from antibodies to reactive thiols in albumin, free cysteine or glutathione in the plasma. This maleimide exchange happened on the highly solvent-accessible site. On the contrary, the conjugation site partially accessible and surrounded by positively charged amino acid residues underwent succinimide ring hydrolysis rapidly, which stabilized the linker by protecting it from maleimide exchange. ADCs with rapid drug loss due to maleimide exchange showed decreased therapeutic activity in mouse xenograft model experiments and increased liver toxicity in the safety study in rats. The phenomenon was also observed in thiol-reactive linkers with different design at drug sites [24].
Application of the click chemistry and unnatural amino acids on site-specific antibody conjugation
Click chemistry is a modular synthetic concept to the assembly of new molecular entities quickly and reliably by joining small units together [25]. Its excellent features - biocompatible, highly specific, fast - makes it widely used in bioconjugation, including protein labeling in vitro and in vivo, labeling of living cells, and assembling of glycans, etc. Azide alkyne Huisgen cycloaddition which is the copper-catalyzed Huisgen 1,3-dipolar cycloaddition between azides and terminal alkynes is the most representative and commonly applied click chemistry. Other reactions such as saudinger ligation and oxime ligation are also used in bioconjugation [26].
To label proteins in vitro or in vivo, unnatural amino acids, with alkynyl or azido group or other functionalities for click, need to be incorporated to provide sites for modification [27]. The most successful approach to genetically incorporating unnatural amino acids is to reassign the amber codon, the least frequently used stop codon, to the unnatural amino acid of interest, and introduce an exogenous aaRS/tRNA pair that is specific to the unnatural amino acid and its reassigned codon but orthogonal to the host cell to enable the translation of the amber codon into the unnatural amino acid. The aaRS/tRNA pair should be evolved, by several rounds of positive/negative selection using an aaRS (aminoacyl-tRNA synthetase) mutant library with its cognate tRNA, to ensure the introduced aaRS/tRNA pair only responds to the amber codon [28-29]. Then the protein can be modified sitespecifically through the click reaction.
The first application of above approach to generate sitespecific ADCs was reported by Jun et al. [30] recently (Figure 3).
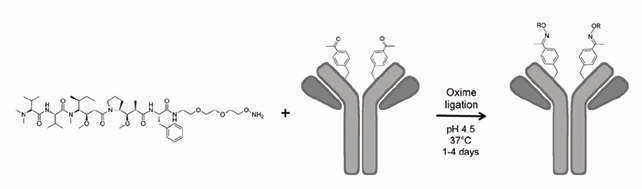
Figure 3:This figure is adapted from [30]. An unnatural amino acid p-Acetylphenylalanine (pA Illustration of a site-specific ADC generated by Jun et al [30]. cPhe) carrying a keto group was site-specifically incorporated into the anti-Her2 antibody trastuzumab. An auristatin derivative with a non-cleavable linker containing a terminal alkoxy-amine group was conjugated to the keto group in the antibody by forming a stable oxime bond.
An unnatural amino acid p-Acetylphenylalanine (pAcPhe) carrying a keto group was site-specifically incorporated into the anti-Her2 antibody trastuzumab both in E. coli and in CHO cell line. An auristatin derivative with a non-cleavable linker containing a terminal alkoxy-amine group was conjugated to the keto group in the antibody by forming a stable oxime bond. Both in vitro and in vivo studies showed that the new site-specific ADC is excellent in stability, pharmacokinetics and efficacy. Though it is not easy to genetically manipulate the protein translation process in mammalian cells, this engineered CHO cell still achieved high expression yield, implying its application value.
CONCLUSIONS
In order to generate ADCs with high selectivity and potency, continuous efforts have been devoted to exploring new antibody targets and formats, developing highly potent cytotoxic drugs, and optimizing the linkage between the antibody and the drug. Safety and efficacy of ADCs are greatly influenced by their pharmacokinetics, in which the linkage plays a critical role. At present, peptide linkers and non-cleavable linkers are regard to be the most successful linker design in the drug side and the exploration of other drug release mechanisms is also underway. Site-specific ADCs, which is highly homogenous both for conjugation site and stoichiometry of payload, is a big advance in the development of ADCs. They are superior to conventional ADCs due to the improved safety and efficacy displayed in preclinic studies. The application of site-specific protein modification technology has greatly improved the development of site-specific ADCs. The first site-specific ADC was generated by site-specifically incorporating cysteines, with which drugs are conjugated through thiol-reactive linkers. Recent progress was made by incorporating unnatural amino acids carrying functional groups for site-specific conjugation by click chemistry. Advances in molecular biology and chemical biology will benefit searches on ADCs, generating new ADC with better safety and efficacy that are highly desirable in cancer immunotherapy.
ACKNOWLEDGEMENTS
We would like to thank Prof. Jianxin Ji and Prof. Zhi-Xiong Jim Xiao for helpful discussions.
REFERENCES
2. enter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009; 13: 235-244.
6. Trail P.A., Antibody Drug Conjugates as Cancer Therapeutics. Antibodies, 2013, 2: p. 113-129.
9. amle NK. Antibody-drug conjugates ace the tolerability test. Nat Biotechnol. 2008; 26: 884-885.
10. eicher BA. Antibody-drug conjugate targets. Curr Cancer Drug Targets. 2009; 9: 982-1004.









































































































































{kind=link}