Function of 2-Arachidonoylglycerol Hydrolyzing Enzymes in Brain Development and Neurodegenerative Disease
- 1. Department of Pathology, Institute for Developmental Research, Japan
Abstract
Monoacylglycerol lipase (MAGL) and a/b hydrolase domain-containing protein (ABHD) have been recently identified as key enzymes in the hydrolysis of 2-arachidonoylglycerol (2-AG), one of the endocannabinoids (eCBs) in the brain. Brain MAGL generates free fatty acids such as arachidonic acid, which regulate proinflammatory cytokine production or termination of eCB-mediated synaptic retrograde signaling. It has been elucidated that dysfunction of MAGL and eCB metabolism are involved in several neurodegeration models via distinct mechanisms. ABHD6 is also important for tuning of 2-AG signaling in synaptic function. ABHD12 encodes serine hydrolase, mutations in which cause the neurodegenerative disorder PHARC (polyneuropathy, hearing loss, ataxia, retinosis pigmentosa, and cataract), and is involved in microglial homeostasis. Recent advances in our understanding of these eCB-metabolizing enzymes have complemented existing studies of eCB receptors in development and neurodegeneration models.
Keywords
ABHD; CB receptor; MAGL; Neurodegeneration; Serine hydrolase.
CITATION
Kouchi Z (2016) Function of 2-Arachidonoylglycerol Hydrolyzing Enzymes in Brain Development and Neurodegenerative Disease. JSM Cell Dev Biol 4(1): 1017.
ABBREVIATIONS
ABHD: α/β Hydrolase Domain-Containing Protein; AD: Alzheimer’s Disease; AEA: N-Arachidonoylethanolamide; 2-AG: 2-Arachidonoylglycerol; CBR: Cannabinoid Receptor; DAGL: Diacylglycerol Lipase; eCB: Endocannabinoid; MAGL: Monoacylglycrol Lipase
INTRODUCTION
Monoacylglycerol lipase (MAGL) was originally identified as a soluble hydrolase that degrades monoglycerides, producing fatty acids and glycerol in the last step of the sequential hydrolysis of triglycerides [1]. MAGL is ubiquitously distributed in most of tissues including brain and immune cells. Structurally, it possesses a catalytic triad in its sequence formed by Ser122, Asp239, and His269 with a unique α/β-hydrolase fold, and is therefore known as a serine hydrolase [2]. ABHD6 and ABHD12 are predicted to be integral membrane proteins and also categorized as serine hydrolases [3]. Brain MAGL and ABHD6 are specifically known to hydrolyze 2-AGs, one of the 2-acylglycerol, catalytic products of either of the two diacylglycerol lipases (DAGLα or DAGLβ). Both serine hydrolases are required for spatiotemporal termination of 2-AG mediated signaling initiated by its cannabinoid receptors (CB1R or CB2R) in neuronal or immune function.
In neurons, MAGL is localized to presynaptic terminals and regulates the amounts of postsynaptically synthesized 2-AG, which activates presynaptic CB1R, resulting in modulation of excitatory or inhibitory neurotransmitter release known as depolarization induced suppression of excitation (DSE) and inhibition (DSI) [4]. As a hub for the signaling involved in neuroinflammation, 2-AG metabolism and its signaling through cannabinoid receptors (CBR) have also been extensively studied in neurodegenerative disease models such as Alzheimer’s disease (AD), Parkinson disease (PD), multiple sclerosis, stroke, and Down syndrome [5-8]. Notably, CB2R expression is hardly detectable in healthy conditions but is induced in immune cells including microglia or myeloid progenitor cells upon inflammation, and its activation suppresses axonal loss and microglial neurotoxicity in several neurodegeneration models [9]. Since modulation of eCB signaling by neuronal CB1R causes several unwanted psychotrophic effects, development of a CB2R-specific reagent has become an attractive strategy in intervenion for neuroinflammation from a clinical perspective.
Recent studies have shown that MAGL has a proinflammatory role by generating arachidonic acid through 2-AG hydrolysis in several neurodegeneration models [10-12]. This series of studies was initiated by the unexpected findings that inactivation of MAGL by genetic and pharmacological methods caused significant reduction in AA levels with attenuation of cytokine productions in the brain, although cytosolic phospholipase A2 (cPLA2)-deficient mice did not exhibit altered levels in lipopoly-saccharide (LPS) treated mice or Parkinson mouse models [10]. MAGL inhibition also causes prevention of neuroinflammation and amyloid β accumulation through suppression of prostaglandin metabolism rather than the CB1R or CB2R signaling axis in a mouse model of AD [11,12]. Microglial MAGL function itself is not sufficient for intrinsic production of inflammatory cytokines, so its activity seems to be subjected to complex regulation in neuron-glia networks in neuroinflammation [13,14]. MAGL inhibitors are attractive candidates for a potential strategy in next generation treatments to combat a broad range of neurodegenerative or neuropsychiatric diseases. In this review, recent progress in our understanding of the activation mechanisms of endocannabinoid degrading enzymes and their functions in neuropathology are discussed with reference to several disease models.
Signaling pathway of MAGL and other 2-AG hydrolyzing enzymes
Endocannabinoids (eCBs) are generated by both neurons and glia in the brain. The primary bioactive eCBs in the brain are 2-AG and N-arachidonoylethanolamide (AEA), and although 2-AG has a lower affinity than AEA to CB1R or CB2R, it is a more effective agonist to the CBR than AEA [15]. 2-AG, the most abundant eCB, is mainly generated by sequential hydrolysis reaction of phosphoinositiol lipids such as PIP2 by PI-specific phospholipase C (PI-PLC) and diacylglycerol lipase (DAGL) when ionotrophic or metabotropic receptors are activated by ATP or glutamate following depolarization (Figure 1) [16-19].
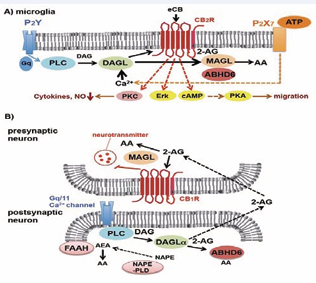
Figure 1: Schematic depiction of enzymatic steps regulating 2-AG production and degradation in microglia (A) and retrograde signaling in the nervous system (B). (A) In microglia, ATP-dependent activation of purinergic receptors causes sequential activation of the 2-AG generation system catalyzed by PLC and DAGL, although basal 2-AG production is retained by other mechanisms. eCB-mediated CB2R signaling activates microglial migration through Erk and PKA, and suppresses cytokine induction by PKC. (B) In neurons, AEA, a substrate of FAAH, is synthesized from N-acyl phosphatidylethanolamine (NAPE) by NAPE-PLD but this step also seems to be mediated by other unknown catalytic reactions including PLA or PLD. 2-AG is generated from phosphatidylinositol lipid precursor by the sequential reactions of PLC and DAGLα or DAGLβ. DAGLα is the primary hydrolyzing enzyme for 2-AG production. Postsynaptically generated eCBs traverse the synaptic cleft and activate CB1R localized at presynaptic compartments, resulting in the inhibition of neurotransmitter release. ABHD6 terminates 2-AG signaling at postsynaptic compartments, whereas MAGL hydrolyzes the lipophilic 2-AG in presynaptic regions.
These signaling cascades are required for microglial activation or retrograde signaling at synaptic transmission in excitatory or inhibitory neurons; however, 2-AG homeostasis is known to be attenuated by inflammatory conditions accompanied with axonal damage such as experimental autoimmune encephalomyelitis [21]. 2-AG production is normally stimulated by neuronal depolarization or G protein-coupled-receptor activation as described, whereas differing pathways are also involved in basal 2-AG synthesis as shown by Gq/G11 knockout mice [22]. 2-AG hydrolysis is dynamically balanced by Ser hydrolases, cyclooxygenases, lipooxygenases or fatty acid amide hydrolase (FAAH) in the brain, and 2-AG activates its receptors CB1R and CB2R in neurons and immune cells under the regulation of multiple degradation systems [23]. FAAH preferentially hydrolyzes AEA, whereas MAGL specifically hydrolyzes 2-AG in the brain. MAGL colocalizes with CB1R at presynaptic terminals in neurons, reflecting the fact that MAGL specifically terminates CB signaling through 2-AG degradation regardless of the source of the eCB [3].
Brain MAGL hydrolyzes 85% of 2-AG and other brain hydrolases, including ABHD6 and FAAH, metabolize residual amounts of 2-AG under normal conditions [24]. MAGL has several splicing isoforms in various tissues and its expression is spatiotemporally regulated by proteolytic degradation in neurons and microglia [2, 13, 23, 25-27]. This complexity of the MAGL expression system may be utilized for spatiotemporal dynamics of 2-AG signaling in brain development and neuroinflammatory conditions (Figure 2).
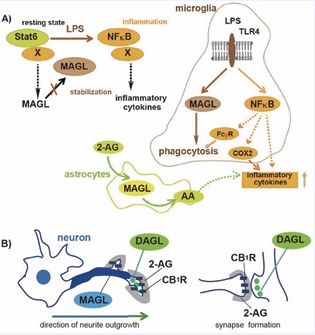
Figure 2: Regulation of MAGL expression and its function in microglia (A) and neuronal outgrowth (B). (A) Microglial MAGL expression is maintained by Stat6-mediated transcription in the resting state, whereas LPS treatment downregulates MAGL transcription but restores its stability by inhibiting unknown proteolytic mechanisms. MAGL promotes FcγR-mediated phagocytosis but not induction of inflammatory cytokines mediated by NFκB. MAGL is required for microglial activation and production of inflammatory cytokines through 2-AG degradation and COX-1 dependent generation of proinflammatory upregulation of MAGL. (B) In corticofugal neurons DAGL colocalizes with CB1R in the motile axon tips of growth cones, whereas MAGL accumulates in the axon shafts with its levels decreased towards the motile tip, forming a barrier region to 2-AG responsiveness. As synaptogenesis concludes DAGLα is downregulated and MAGL retains its activity in presynaptic regions.
For example, tuning of 2-AG amounts by local MAGL degradation regulates motility of the growth cone and axonal wiring during path finding in cortical neurons [25]. Similar implications for MAGL protein turnover have been reported in cholinergic neurons in response to NGF, and breast cancer type 1 susceptibility protein 1 (BRCA1) functions as an E3 ubiquitin ligase for its downregulation to activate 2-AG/CB1R signaling in the motile growth cone [26]. Interestingly, in many patients with schizophrenia, the expression levels of neuregulin-1 and its receptor ErbB tyrosine kinases are known to be altered in GABAergic neurons, and chronic treatment with neuregulin-1 increases MAGL expression, resulting in decreased 2-AG effects on long-term depression of inhibitory synapses [27]. MAGL expression also seems to be also dynamically regulated during neuroinflammation. Post-mortem analysis of the hippocampus in Alzheimer’s patients has shown that MAGL accumulates in IBA-1 positive microglia around senile plaques, and progressive lipid mediator prostaglandins expression in activated microglia and astrocytes is highly correlated to the disease progression state [28]. In microglia, MAGL transcription is mostly regulated by Stat6 but is also subjected to proteolytic degradation, and LPS treatment facilitates its stability by an unknown mechanism [13]. Thus, in situ MAGL function might be altered by posttranslational modification in neuroinflammatory conditions with disruption of the homeostasis of 2-AG production.
ABHD6 is capable of degrading 2-AG as Ser hydrolase, and it has a predominant role in 2-AG hydrolysis in BV-2 cells, which lack MAGL expression [29-31]. ABHD6 also controls 2-AG metabolism at postsynaptic compartments in electronically stimulated neuron, which is distinct from that MAGL is localized at presynaptic regions in charge of bulk 2-AG degradation. Therefore, the cell-intrinsic activity of ABHD6 allows its tuning at lower levels than MAGL-mediated catalysis in accordance with 2-AG synthesis. In a neuropathologic model ABHD6 blockade with the specific inhibitor WWL123 reduced epileptiform seizures provoked by pentylentetrazole independent of CB1R and CB2R signaling [32].
Another Ser hydrolase, ABHD12, was initially categorized as a 2-AG hydrolyzing enzyme in the brain by the FP-biotin labeling method termed activity-based protein-multidimensional protein identification technology (ABPP-MudPIT) in brain membranes [24]. ABHD12 was identified as a hereditary causative gene of the neurodegenerative disease PHARC and patients with ABHD12 mutations were identified initially as phenocopies of Refsum disease, in which phytanic acid metabolism is dysregulated [33,34]. Despite the fact that mammalian cells expressing exogenous ABHD12 possess 2-AG hydrolyzing activity in vitro, ABHD12 knockout mice show significant increases in lysophosphatidylserine (lysoPS) but not monoacylglycerol, indicating that ABHD12 metabolizes lysoPS in vivo rather than 2-AG in the brain [35, 36]. Furthermore, ABHD16a has recently been identified as a phosphatidylserine hydrolyzing enzyme generating its substrate lysoPS, and interplay between ABHD12 and ABHD16a is considered to be important for phenotypic conversion of microglia and macrophages [37].
Endocannabinoid metabolism in the immune system during neuroinflammation
The neuroprotective roles of eCB signaling in neuroinflammation have also been extensively studied in microglia, and eCBs are known to reduce the expression levels of interleukin 6 (IL-6), interleukin 1 (IL-1), and tumor necrosis factor alpha (TNFα) in microglia [38]. However, neither CB1R nor CB2R antagonists affected CB1R agonist WIN55, 212-2-mediated effects, suggesting that other unknown eCB responsive receptors or eCB metabolizing enzymes are implicated in cytokine regulation. Noticeably, Cravatt’s group has indicated that brain MAGL inactivation nullified microglial activation and cytokine production through downregulation of arachidonic acid and prostaglandins in LPS or MPTP (1-methyl-4-phenyl 1,2,3,6-tetrahydropyridine)-injected mouse models by liquid chromatography-mass spectrometry analysis [10]. However, inactivation of microglial MAGL by either small interfering RNA-mediated knockdown or pharmacological inhibition by its specific inhibitor JZL-184, did not affect cytokine induction, and exogenous expression of MAGL in BV-2 microglial cells did not promote their induction by LPS treatment, although 2-AG hydrolyzing activity was enhanced by MAGL expression [13]. Viader et al. recently showed that astrocytic MAGL is responsible for the coordinated generation of AA and prostaglandins from 2-AG in neuroinflammatory conditions by using MAGL conditional knockout mice [14]. These findings indicate that 2-AG homeostasis may be balanced by neuronal and astrocytic MAGL, and generated AA might be utilized in diverse signaling pathways to stimulate cytokine production in inflammatory conditions in neuronal/glia networks including microglia.
Although astrocytic MAGL are important for the sources of the proinflammatory lipid mediator AA, CB2R signaling is an important avenue for microglial functions. Recruitment of activated microglia to dying neurons is the initial response in neuroinflammation. In pathological situations activated by ATP with neuronal injury, 2-AG upregulates microglial cell motility through CB2R localized at lamellipodia tips [39]. Microglial CB2R activation by its agonist AM1241 is also required for attenuation of its inflammatory phenotypic conversion and maintenance of the alternative activated form when treated with LPS and interferon gamma (IFNγ) [40]. Thus, CB2R signaling may regulate microglial differentiation during inflammation. Although it remains unknown whether microglial differentiation is involved or not MAGL is shown to promote Fcγ receptor-mediated phagocytosis by using primary microglia and BV-2 cell lines [13].
In AD models, local eCB production in the brain seems to contribute to cognitive impairment. Amnesia caused by the Aβ peptide is prevented by treatment with a CB1R antagonist, whereas intracellular CB1R supports lysosomal stabilization by preventing Aβ-induced neurotoxicity accompanied with excess membrane permeability, suggesting that spatiotemporal regulation of CB1 signaling may be important for neuroprotection during AD onset [41,42]. As pathological deterioration progresses, 2-AG dynamics mediated CB2R signaling have important roles in both brain microglia and peripheral immune cells. For example, CB2R and FAAH are selectively expressed in astrocytes and microglia surrounding neuritic plaques in postmortem brain of AD patients [43]. In an amyloid precursor protein (APP) mouse model with Aβ administration, activation of CB2R with WIN 55, 212-2 suppressed microglial activation and neurotoxic effects in vivo [44]. CB2R activation in parenchymal microglia in a Huntington’s disease (HD) model is also known to attenuate motor deficits and neuroinflammation to prevent astrogliosis and striatal excitotoxicity [45]. In addition, CB2R stimulation in peripheral immune cells has been shown to be important for slowly progressing models of HD such as BACHD or R6/2 mice by the combined use of its agonist GW405833 and its plasma selective antagonist SR14428 [46]. When neuropathological symptoms are associated with a strong inflammatory reaction and immune cell infiltration in the brain like multiple sclerosis, CB2R expression is significantly increased in microglia and bone-marrow myeloid progenitor cells, and its loss of function causes severe clinical deterioration by hampering peripheral cell trafficking and preventing microglial recruitment to inflammatory lesions due to decreases in chemokine ligands or receptors [47].
Function of MAGL and CB receptors in developing brain and neurodegenerative disease
An imbalance in eCB signaling is known to cause developmental abnormality as shown by deficits in normal proliferation of pyramidal cell progenitors and proper axon fasciculation in CB1R knockout mice [48]. Harkany’s group have shown that MAGL has an important role in temporal establishment of spatial restriction for the signal competence for 2-AG with the DAGL and CB1R signaling axis during corticofugal axonal growth [25]. Axonal pathfinding and differentiation of oligodendrocytes in corpus callosum are also regulated by MAGL activity in CB1R in coritcofugal neurons and CB2R expressing oligodendrocytes by spatiotemporal inactivation of Slit/Robo1/2 [49]. Interestingly, CB1R function is important for inhibitory control of pyramidal cells in specific layers of the cortex through intracortical or corticostriatal processing of neuronal activities and specification of subcortical projections regulating corticospinal motor function [50,51]. The importance of developmental 2-AG regulation by MAGL and DAGLα has been implicated in cholinergic innervation of CA1 pyramidal cells during the fetal development of the hippocampus [26,52]. Thus, eCB turnover and CBR function are tightly regulated by coupling with spatial MAGL regulation during brain development (Figure 2).
In contrast, the importance of MAGL as a signaling hub independent of CBR function has emerged from several neuropathological studies. Genetic ablation of MAGL in PS1/ APP transgenic mice suppresses amyloid plaque formation with attenuation of gliosis and microglial activation [11]. In addition, treatment of these mice with the MAGL-specific inhibitor JZL184 decreased beta-secretase 1 (BACE1) expressionand Aβ production in both the cortex and hippocampus, and inhibit AA and prostaglandin production, resulting in improvement of synaptic plasticity and spatial learning and memory [11,12]. Clinically, inhibitors of cyclooxygenase-1 (COX-1) and COX-2 catalyzing AA oxidation are not approved for chronic use as AD treatments due to side effects such as hemorrhage [52,53]. Therefore, MAGL inhibitors are one of the most attractive treatment strategies, but the signaling mechanisms of MAGL in relation to neurotoxicity remain elusive. Subsequent studies have shown that 2-AG accumulation by MAGL inhibition upregulates miR-188-3p through peroxisome proliferator-activated receptor gamma (PPARγ) independent of CB1R or CB2R, and miR-188-3p reduces BACE1 and Aβ formation, therefore improved synaptic structure and function by MAGL suppression may be regulated by multiple neuroprotective signaling cascades [54]. Similar neuroinflammatory effects of MAGL without CBR agonism are indicated in a chronic MPTP mouse model with JZL-184 administration [55].
Interestingly, people with Down syndrome (DS) caused by triplication of chromosome 21 are known to develop AD-like pathology such as deficient cognition due to dysfunction in the prefrontal cortex during aging. Pharmacological inhibition of MAGL by JZL-184 ameliorates the long-term memory defects and enhanced spontaneous locomotor activity observed in Ts65Dn mice, a genetic DS mouse model [8]. Dyrk1a, a mammalian ortholog of Drosophila minibrain, is within the critical region of chromosome 21q22 closely associated with DS. Overexpression of Dyrk1a in mice increased the number of spines and defects in eCB-mediated long-term depression (eCB-LTD) in pyramidal neurons; however, JZL-184 treatment restored normal eCB LTD and prefrontal cortical function in TG mice [56]. These beneficial effects of MAGL inhibition, presumably coupled with CBR signaling, are only observed in the pathological state, with no overt effect on healthy conditions, suggesting a specific role of MAGL in the neurological deterioration.
Recent studies have shown that MAGL plays an important role in depression and chronic stress conditions. Chronic unpredictable mild stress to mice induces depressive-like behavior due to impairment of DSI in hippocampal CA1 pyramidal neurons and JZL-184 treatment rescues the 2-AG-mediated retrograde synaptic depression [57]. This mode of depression causes inactivation of mammalian target of rapamycin (mTOR) in the hippocampus, whereas chronic JZL-184 treatment ameliorates symptoms through restoration of the CB1R/mTOR activation pathway. Chronic MAGL inactivation has also been shown to rescue decreases in long-term potentiation (LTP) and neurogenesis in the dentate gyrus observed in mice subjected to unpredictable mild stress [58].
CONCLUSION
MAGL, FAAH, and ABHD6 have recently been identified as critical regulators of eCB metabolism including 2-AG, and spatially regulate the function of eCB receptors in distinct signaling networks to maintaining eCB homeostasis during brain development. Their organized regulation is disturbed in neuroinflammatory environments as seen in changes in DAGL, MAGL or ABHD6 activities and their expression in several neurodegeneration disease models [28,43]. Presumably, neuronal or glial damage may alter their subcellular localization, resulting in disruption of 2-AG dynamics that are detrimental to synaptic function and homeostasis regulated by microglia. The highly selective MAGL inhibitor, JZL-184, has been widely used for assessment of its function in combination with genetic MAGL-null mice, and its neurological differential effects have also been indicated in chronic effects on the desensitization of CB1R [59]. Recently, potent ABHD6 and ABHD12 inhibitors have been developed [29,35]. Designing more specific chemical compounds would be useful for elucidating the unknown functions of these serine hydrolases in the brain, and for the development of promising therapeutic strategies in several neuropathologies.









































































































































{kind=link}