Bile Acids Induce Intestinal Metaplasia, Gastric Cancer, and Chemoprevention
- 1. Board-certified Gastroenterologist in Los Angeles, USA
Abstract
The symptoms of Gastric Cancer (GC) are often nonspecific, which can delay diagnosis and lead to a poor prognosis. In Intestinal Metaplasia (IM), bile reflux (BR) replaces the gastric mucosal epithelium with intestinal epithelium. Chenodeoxycholic Acid (CDCA) and Deoxycholic Acid (DCA), among the Bile Acids (BAs) in BR fluid, can stimulate BA receptors in the stomach. CDCA activates FXR, while DCA activates both GPCRs and FXR. DCA, a significant risk factor for gastrointestinal cancer, damages gastric epithelial cells by disrupting the outer mitochondrial membrane. This disruption produces reactive oxygen and nitrogen species, leading to DNA and chromosomal damage. Additionally, DCA causes gastric epithelial cells to progress from complete IM to incomplete IM. DCA activates multiple oncogenes, especially β-catenin, contributing to gastric carcinogenesis. In patients with GC, DCA increases levels of Dickkopf-1 (DKK1), which plays a crucial role in epithelial-mesenchymal transition (EMT), immune evasion, resistance to chemotherapy and immunotherapy, and metastasis by activating the β-catenin/Myc/STAT3 pathways. The vitamin D receptor (VTDR) regulates anti-cancer actions by halting cell growth, promoting apoptosis and autophagy, preventing blood vessel formation, and modulating immune responses and metastasis in GC through the inhibition of β-catenin.
Keywords
• Bile Acid; Gastric Cancer; Vitamin D
Citation
Rheem D (2025) Bile Acids Induce Intestinal Metaplasia, Gastric Cancer, and Chemoprevention. J Cancer Biol Res 12(1): 1151.
INTRODUCTION
In 2020, over one million cases of gastric cancer were reported, leading to approximately 769,000 deaths, making it the fifth most common cancer worldwide by incidence and the fourth leading cause of cancer-related death [1]. The etiology of GC is highly complex, involving intricate interactions among various risk factors in patients. Factors such as HP infection, age, male gender, smoking, and alcohol consumption collectively contribute to the development of GC [2].
GC is difficult to prevent and treat due to limited understanding of its pathogenesis and challenges in early diagnosis. Various treatments, including surgery, chemotherapy, radiotherapy, and immunotherapy, are used for GC, but endoscopic and surgical removal remains the main options for early-stage cases. Although progress has been made in understanding the pathogenesis and treatment of advanced gastric cancer, the 5-year survival rate is <10% [3]. Most patients with GC are diagnosed at an advanced stage. Therefore, researching GC development and understanding the underlying molecular mechanisms are vital for developing the most effective prevention and treatment strategies to lower the high rates of morbidity and mortality associated with GC. The progression of gastric cancer involves several stages, such as chronic gastritis, atrophic gastritis (AG), intestinal metaplasia (IM), dysplasia, and ultimately the development of GC. Among these, IM is considered a precursor to GC [4]. Chemoprevention is a strategy aimed at preventing or delaying the onset of GC. The roles of VTD and statins are discussed.
Atrophic gastritis and HP
AG typically develops after several decades of chronic Helicobacter pylori (HP) infection. The loss of gastric mucosal glands begins in the antrum and spreads to the body. Another factor contributing to this process is an autoimmune response that targets parietal cells or their components. Most cases of gastric atrophy are caused by chronic HP infection, with 89% of non-cardia GCs and 78% of all GC cases resulting from these infections. Numerous studies continue to emphasize the role of HP in the development of GC. HP infection usually occurs during childhood, but GC diagnosis often happens five or more decades later, highlighting a significant age gap. Most GC cases develop in individuals with IM in their antrum. The antrum presents challenges for HP survival in GC patients due to decreased nutrition caused by gastric mucosal atrophy and cytotoxic bile. Chronic AG can be effectively induced in rats using deoxycholate (DCA) [5]. Eradicating HP reduces the risk of GC for patients without IM.
However, it does not stop progression in patients with IM or dysplasia, indicating a “point of no return” [4-7]. After eradication, the absence of HP does not prevent cytotoxic BR from promoting GC. These observations highlight the importance of factors beyond HP in determining GC risk. This emphasizes the need for a more comprehensive understanding of the molecular pathogenesis of GC.
Intestinal Metaplasia
HP has long been recognized as a significant factor in the development of IM. However, recent research challenges this view, suggesting that cytotoxic bile reflux, rather than HP, triggers the transition from normal gastric epithelial cells to IM [8]. According to a large, multi-center, cross-sectional study, the risk of developing IM is significantly higher in patients with elevated bile acid levels, regardless of their HP infection status [9]. DCA may promote gastric IM by activating the IL-6/STAT3 pathway [10]. The synthesis of primary BAs, specifically cholic acid (CA) and CDCA, occurs in the liver from cholesterol. Most conjugated primary BAs are reabsorbed in the terminal ileum, with a small portion entering the colon. There, colonic bacteria facilitate deconjugation and dehydroxylation, producing secondary BAs, namely DCA and lithocholic acid (LCA) [6].
CA, CDCA, and DCA are reabsorbed in the small and large intestines and transported back to the liver, while most LCA is excreted in feces. Reports indicate that the hepatic fractional uptake of CA is approximately 90%, whereas the uptake of CDCA and DCA ranges from 70% to 80% [8]. Due to its hydrophobic nature, LCA is less readily absorbed by the colon and is mainly excreted in the stool. The enterohepatic circulating BA pool in adult humans consists of 30–40% CA, 30–40% CDCA, 30% DCA, and 5% LCA [11] (Figure 1). In patients with uncontrolled high cholesterol and low vitamin D, DCA levels may be further elevated in the enterohepatic BA pool. Bile reflux is closely associated with the development of IM and GC [7-9]. Gastric IM involves the transformation of the gastric mucosa into small and large intestinal mucosa due to environmental stress, with BAs playing a crucial role. The gastric mucosa adapts to withstand a harsh, acidic environment. When cytotoxic and hydrophobic BAs reflux from the duodenum into the stomach, the gastric mucosa transforms into intestinal epithelium. GIM refers to the conversion of differentiated gastric epithelial cells into a distinct cell type found in the small or large intestine [5]. This IM results from reprogramming gastric stem cells into intestinal stem cells.
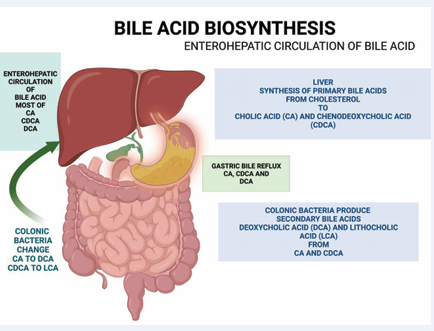
Figure 1: The liver cells synthesize the Bile Acids (BAs) by oxidation of cholesterol, which are Cholic Acid (CA) and Chenodeoxycholic Acid (CDCA). Most BAs are reabsorbed at the terminal ileum. However, some BAs are spilled over the large intestine, and colonic bacteria uncouple and convert primary BAs into secondary BAs: Deoxycholic Acid (DCA) and Lithocholic Acid (LCA). LCA is too hydrophobic, and only a small amount of it can be reabsorbed.
The BR of two hydrophobic bile acids, CDCA and DCA, is linked to complete intestinal metaplasia (CIM) (type I), incomplete intestinal metaplasia (IIM) (types II or III), and gastric cancer (GC). The gastric mucosa progresses to a fully developed form of IM, characterized by a small intestinal epithelium with well-formed goblet cells and columnar absorptive cells. The IIM mucosa lacks a brush border and does not contain goblet or columnar cells that secrete mucin. A high iron diamine stain distinguishes sialomucins—appearing blue in the small intestine and colon (type II IM)—from sulfomucins, which appear brown in the colon (type III IM). Incomplete type III is more closely associated with GC (incomplete type III > incomplete type II > complete type I) [12]. In younger patients, BR mainly consists of CDCA. The gastric mucosa develops into a complete form of IM, characterized by small intestinal epithelium made up of well-developed goblet cells and columnar absorptive cells. In older patients, the level of DCA increases with age, contributing to its accumulation. Elevated DCA levels can trigger IIM and promote the development of GC, mainly through activating GPCRs, the epidermal growth factor receptor (EGFR), and the β-catenin pathway [13]. GC exhibits heterogeneity, with various DNA and chromosomal changes. Different stages of carcinogenesis involve activating specific oncogenes and suppressing tumor suppressor genes. DCA also increases the production of DKK1 in advanced GC [14] (Figure 2).
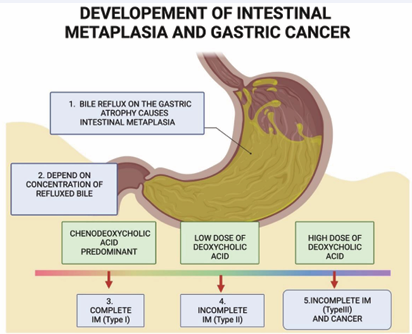
Figure 2: Bile reflux of hydrophobic Bile Acids (BAs) causes Intestinal Metaplasia (IM) in atrophic gastritis patients. Farnesoid X receptors (FXRs) reduce BA production in the liver and detoxify the hydrophobic BAs. CDCA can activate FXR. When patients are young, most of the BAs are primary bile acids. DCA can be reabsorbed by passive diffusion and returned to the enterohepatic BA pool. As the patient ages and consumes a high-fat, low-fiber diet, the patient’s BA pool will be replaced with DCA. DCA activates GPCRs to induce incomplete IM and gastric cancer.
The risk of GC in patients with IIM was 5.16 times higher (95% CI, 3.28-8.12), compared to those with IM. Additionally, the risk of GC associated with type III IM was the highest among incomplete type II, showing an increased risk of 2.88 times (95% CI, 1.37-6.04) [15]. Within the IM groups, factors such as bile reflux grades, age, high-fat diet, and a family history of GC were identified as independent risk factors for IM and for progression from IM to GC [16].
The effect of BAs on GIM formation was studied using human gastric biopsy tissues, revealing higher levels of CDX2, MUC2, and Farnesoid X receptor (FXR) [17]. CDCA, a primary BA, activates FXR in complete IM, leading to increased CDX2 expression. CDX2 is an intestine-specific transcription factor found from the duodenum to the rectum but absent in the gastric mucosa. FXR promotes the binding of p50 to the CDX2 promoter and enhances MUC2 transactivation. BAs stimulate FXR in this order: CDCA > DCA > LCA > CA [18]. FXR signalling can reduce the proliferation of Leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5) stem cells, inhibit tumor growth, and support intestinal health by restoring the intestinal barrier [19]. The BA pool primarily consists of primary BAs in young patients, with CDCA activating FXR to replace the gastric mucosa in complete IM. As patients age and adopt a high-fat diet, DCA levels in the BA pool increase. DCA activates GPCRs, leading to IIM and GC.
Treatment with 200 µM/L of DCA raised Takeda G protein-coupled receptor 5 (TGR5) levels in IM samples. TGR5, also called G-protein-coupled receptor 1 (GPBAR1), mediates the DCA-induced metaplastic phenotype. Mechanistically, DCA treatment increases hepatocyte nuclear factor 4α (HNF4α) expression through GPBAR1, thereby activating the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway [20].
Secondary BAs mainly activate GPCRs, ranked by effectiveness: LCA > DCA > CDCA > CA [19]. Treatment with 400 µM/L of DCA increased the expression of IM-related genes CDX2 and MUC2. DCA reflux triggers IM in the stomach, which could lead to GC [10]. The plasma DCA level in cholecystectomy patients and mice was notably higher than in sham-operated mice. The tumor burden in these patients and mice increased significantly due to activation of the DCA/β-catenin/LEF/TCF/Myc pathway [21]. BAs were found to increase intestinal marker expression in gastric epithelial cells while downregulating DKK1. Both mRNA and protein levels of DKK1 were lower in GIM tissues. Interestingly, DKK1 promoter methylation was higher in GIM tissues. BAs partly induce methylation of the DKK1 promoter. Promoter methylation and lower DKK1 expression may play a major role in BA-induced GIM development [22]. An analysis of gastric juice from 70 patients with three stages of gastric disease—chronic superficial gastritis, intestinal metaplasia, and gastric cancer—showed differences in the DCA to CA ratio between chronic superficial gastritis and both IM and GC. Colonic bacteria convert CA to DCA, contributing to GC development. Notably, DCA, a key GPCR ligand, accumulates at high levels in individuals with gallstones. Unlike rodents, humans cannot convert DCA to CA in the liver, leading to DCA accumulation in the BA pool [23] (Figure 3).
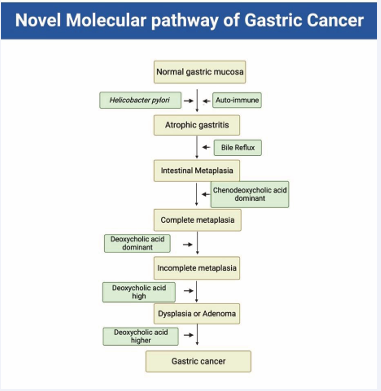
Figure 3: Novel molecular pathway of gastric cancer. Gastric mucosal changes begin from normal gastric mucosa. Helicobacter pylori and autoimmune mechanisms cause atrophic gastritis. Duodeno-gastric bile reflux induces Intestinal Metaplasia (IM). When the patient is young, most of the bile in the stomach consists of primary bile acids (BAs). The primary hydrophobic BA, CDCA, activates the Farnesoid X receptor (FXR), inducing intestinal CDX2. The secondary BA in the stomach is DCA. DCA activates AKT and ERK signaling pathways. DCA in bile reflux alters the gastric mucosa to incomplete IM, dysplasia or adenoma, and eventually GC, depending on the DCA concentration.
High doses of DCA can induce IIM cells to GC cells by activating the β-catenin pathway through EGFR and GPCR, leading to p-AKT activation and GSK-3β inactivation [13]. However, higher doses of DCA are required to activate DKK1 [24].
Gastric carcinogenesis
A diet high in fats and low in fiber leads to increased BA secretion, including both primary and secondary BAs. Secondary BAs (especially DCA) activate cancer-promoting signaling pathways that promote GI cancers in various organs, such as the colorectum, pancreas, liver, bile ducts, and esophagus. DCA is significant in the development of GI cancers [24]. Individuals on a high-fat diet gather DCA in their BA pool at levels three to four times higher than those on a high-fiber diet. In healthy young adults, the gallbladder’s bile usually contains 25% DCA, 35% CDCA, 35% CA, 1% LCA, and 2% UDCA. However, some people have higher DCA levels in their bile. In individuals with cholesterol gallstones, the BA pool is significantly richer in DCA (40.7 ± 9.1% DCA, 25.6% ± 7.1% CDCA, 8.1 ± 3.2% LCA, and 3.3 ± 2.7% CA) [23].
The level of BAs is a key factor in the development of GC in patients’ stomachs. Bernstein et al., showed that adding 0.2% DCA to the diet of wild-type mice for 8-10 months caused colonic tumors in 17 out of 18 mice, including 10 cases of cancer [25]. DCA levels were higher in patients with colorectal cancer (CRC) or adenomas compared to those without these conditions. People on a high-fat diet had fecal DCA levels between 200 and 300 μM, and higher DCA levels were linked to the development of adenomas and CRC. Increased DCA levels activated the EGFR/MAPK signalling pathway. In contrast, people who followed a high-fiber diet had lower DCA levels in their fecal water, reducing the risk of developing colorectal adenomas [26].
DNA, Chromosome Damage, and Anti-Apoptosis
DCA has been linked to damaging DNA and chromosomes by producing reactive oxygen species (ROS) and reactive nitrogen species (RNS) [27]. DCA causes DNA damage in esophageal tumor cells at doses of 100 µM and higher, but not at lower doses due to B activation by DCA. The cellular damage caused by DCA involves disruption of the mitochondrial outer membrane, leading to the production of ROS and RNS, which ultimately result in DNA damage, with double-strand breaks being particularly harmful and potentially leading to mutations and cancer progression [28]. Additionally, DCA impairs several DNA mismatch repair enzymes, causing genome microsatellite instability [29].
Chronic exposure to DCA damages the gastric mucosa and suppresses the expression of proteins related to apoptosis, leading to uncontrolled growth of gastric epithelial cells with DNA and chromosomal damage. The absence of a functional mismatch repair system worsens GC progression [29]. Damage to the centrosome, responsible for producing bipolar mitotic spindles, can cause chromosomal instability (CIN) and aneuploidy. Increased levels of ROS contribute to centrosome amplification, and the loss of p53 may promote this process by elevating polo-like kinase 4, a key protein involved in regulating centrosome number [30]. DCA can induce DNA damage, which may lead to cancer through mutation accumulation. Chromosomal instability (CIN), a major driver of tumor evolution and a hallmark of cancer, results from ongoing errors in chromosome segregation during mitosis [29] (Figure 4).
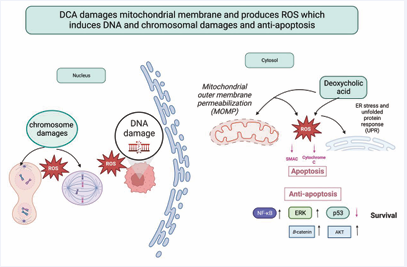
Figure 4: DCA causes DNA and chromosomal damage by disrupting the properties of the mitochondrial outer membrane, which ROS and RNS mediate. DCA can also harm DNA mismatch repair enzymes, such as p53. Additionally, DCA can inhibit apoptosis through pathways involving NF-κB, AKT, ERK, and β-catenin.
THREE MAJOR PATHWAYS IN GASTRIC CARCINOGENESIS
There are many pathways involved in gastric carcinogenesis [31,32]. However, three major pathways will be reviewed: DCA activates three significant cancer-related pathways: G protein-coupled receptors (GPCR), epidermal growth factor receptor (EGFR), and abnormal β-catenin signalling (Figure 5).
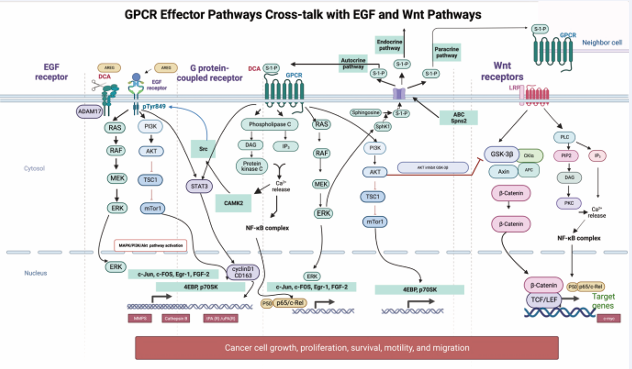
Figure 5: GPCR signaling and its crosstalk with EGF and β-catenin pathways. DCA stimulates GPCRs to activate multiple messenger pathways. DCA-induced CAMKII leads to MAPK activation by recruiting c-Src. DCA promotes the shedding of amphiregulin (AREG) from the membrane receptor ADAM-17. This AREG binds to EGFR and increases p-EGFR and p-Akt levels. SphK1 activates S1P, directing it to the cell periphery. Through Spn2, S1P is transported outside the cell and activates the GPCR itself, as well as neighboring and distant receptors. PI3K promotes GC cell invasion and migration by regulating the AKT/GSK-3β/β-catenin signaling pathway.
EGFR and GPCR.
DCA may cleave disintegrin and metalloproteinase-17 (ADAM 17) to produce amphiregulin, which activates the EGFR signalling pathway. DCA primarily promotes the shedding of amphiregulin (AREG). DCA-induced cleavage of AREG pro-ligand activates EGFR signaling and encourages tumor growth [33]. DCA may activate the EGFR signalling pathway, specifically the Ras/Raf/MEK/ERK/ MAPK pathway, and promote cell proliferation, a well-documented process [34]. Additionally, EGFR can activate the PI3K/Akt/NF-κB pathway, which regulates cell growth and prevents apoptosis [35].
Additionally, DCA activates GPCR through sphingosine 1-phosphate (S1P). S1P is a ligand that activates cell surface S1P receptors 1-5 (S1PR), enabling various pathways that contribute to cancer progression. Sphingosine phosphate kinases (SPHKs) produce S1P, which is exported from cells and activates neighboring or distant S1PRs. These S1PRs, a subset of GPCRs, regulate several signaling pathways involved in carcinogenesis. S1P plays an essential role in carcinogenesis by activating multiple pathways [36]. SPHK phosphorylates sphingosine to generate S1P. In GC, SPHK1 mRNA and protein levels are increased compared to normal gastric epithelial cells. The SPHK1 protein level is higher in GC lesions than in paired adjacent noncancerous tissue [36].
Elevated SPHK1 and GPCR expression play a critical role in determining TNM stages and prognosis in GC patients [36]. The SPHK1 also mediates the migration and invasion of GC cells, involving VEGF, IL-6, and MMP-7 [37]. The increased expression of S1PR1 is strongly linked to shorter overall survival and poor chemotherapy responses in GC patients [38,39].
S1P activates GPCRs linked to the G12/13 family of heterotrimeric G proteins. These GPCRs transmit signals that quickly modify the actin–cytoskeleton by activating the Rho GTPase family, including Rho, Rac, and CDC42. These proteins work together to reshape actin structures and control the cell’s contractile machinery, thereby affecting various cellular processes [39].
Cross-talk between GPCR and EGFR.
DCA-treated cells display intense and sustained activation of ERK1/2 compared to EGF treatment alone. Inhibition of EGFR degradation caused by calcium/ calmodulin-dependent protein kinase II (CAMKII), leads to phosphorylation of EGFR Tyr 84 through the recruitment of c-Src [40]. The combination of DCA and EGF treatment shows synergistic effects. S1P may promote GC progression via Gi- and matrix metalloprotease (MMP)-independent c-Met and EGFR transactivation. However, S1P- or LPA-induced transactivation of HER2 requires MMP activation and the tyrosine kinase activity of EGFR [41]. Furthermore, knocking down the membrane-type bile acid receptor (M-BAR)/TGR5 reduces DCA-induced phosphorylation of EGFR, and DCA transactivates EGFR through M-BAR- and ADAM-EGF-dependent pathways [42].
β-catenin activation, initiation, and cancer stemness in GC
PI3K is activated by stimulated EGFRs or GPCRs, leading to the synthesis of PIP3 and the recruitment of oncogenic effectors, such as the serine/threonine kinase AKT. The AKT allows binding to PIP3, resulting in the accumulation of PDK1 and mTORC2. MTOR is a well-known target of AKT, promoting biosynthetic processes essential for cell growth and proliferation [43].
EGFR and GPCR frequently activate AKT phosphorylation. This phosphorylated AKT inhibits GSK-3β, causing the release of β-catenin from the destruction complex and increasing its levels in the cytoplasm. Consequently, β-catenin accumulates and translocates to the nucleus. In the nucleus, β-catenin binds to TCF/ LEF to promote the expression of Myc, Cyclin D1, and IL-6/STAT3 [14-44]. DCA activates GC cells to promote carcinogenesis by stimulating the PI3K/AKT/β-catenin signaling pathways [14]. VTD suppressed GC cell growth both in vitro and in vivo through downregulating CD44, a transmembrane glycoprotein known as a cancer stem cell marker [45].
GASTRIC CANCER
Early gastric cancer (EGC)
Early gastric cancer (EGC) is defined as a type of GC that is confined to the mucosa and/or submucosa, regardless of lymph node involvement. EGC can be challenging to detect because patients usually do not display typical symptoms such as upper gastrointestinal bleeding, weight loss, vomiting, or abdominal pain unless a national screening program is in place. EGC can be identified when patients with intestinal metaplasia undergo surveillance upper endoscopy. South Korea has been performing biennial endoscopic gastric cancer screening for adults aged 40 and older since 2002. The proportion of early GC increased from 28.6% in 1995 to 63.6% in 2019, and the 5-year survival rates rose from 43.9% (1993-1995) to 77.5% (2015-2019) [7].
Advanced Gastric Cancer (AGC)
When GC cells invade beyond the muscularis propria, it is classified as AGC. Patients presenting with gastric symptoms are often diagnosed at an advanced stage, which may include liver metastasis, obstructive symptoms, ascites, and jaundice. At this stage, these patients are no longer suitable candidates for surgery, and their limited response to chemotherapy and immunotherapy contributes to a high mortality rate. Despite advancements in these treatments, the prognosis for patients with AGC remains poor. A comprehensive retrospective analysis revealed significant differences in median survival rates: patients who underwent surgery and chemotherapy had a median survival of approximately 14.2 months, while those who did not have surgery survived only 7.0 months [7]. Another study showed that the incidence of GC in patients with high bile acid (BA) levels in gastric juice (≥1000 mmol/L) was significantly higher than in those with low gastric BA levels (<1000 mmol/L). A multicenter study indicated that the risk of GC was 2.4 times greater in the high-concentration BA level group compared to other groups [8].
The role of DKK1 in Advanced GC: In patients with IM, precancerous lesions do not display increased levels of Dickkopf-1 (DKK1). DKK1 is epigenetically silenced through promoter methylation [53]. However, serum DKK1 levels in GC patients are significantly higher than in IM cases and healthy controls (all, P < 0.01). DCA is the primary factor leading to DKK1 elevation in GC, as it decreases miR-1 levels. Higher doses of DCA treatment enhance the transcription of SNAI2, which then inhibits the miR-1 promoter. SNAI2 directly binds to the miR-1 promoter, blocking its transcription. DCA lowers miR-1 levels in GC cells, resulting in increased expression of histone deacetylase 6 (HDAC6) and HNF4α compared to IM. HDAC6 epigenetically lifts repression of DKK1 messenger RNA, leading to increased DKK1 levels [7]. Knockdown of circATP8A1 significantly reduces the proliferation and invasion of GC. Exosome-derived circATP8A1 from GC cells promotes M2 macrophage polarization through the circATP8A1/miR-1-3p/STAT6 axis, supporting tumor progression [8].
Serum DKK-1 levels gradually increased as the GC advanced. Additionally, serum DKK-1 levels were significantly higher in patients at TNM stages III and IV compared to those at stages I and II. The level of serum DKK-1 correlated with microvascular invasion, degree of dedifferentiation, and infiltration depth (P < 0.01) [46]. These findings suggest that DKK-1 is an important serological biomarker for carcinogenesis and poor prognosis. Along with increased DKK-1 levels, an increase in β-catenin accumulation was observed in tumor tissues using immunohistochemistry (IHC) staining, which is associated with poorer clinical outcomes [47].
CKAP4 is recognized as a novel receptor for DKK1. The formation of a complex between CKAP4 and phosphatidylinositol 3-kinase (PI3K) ultimately triggers the activation of AKT. The PI3K/AKT signaling pathway is a vital intracellular pathway [48]. When signaling molecules like growth factors, EGF, GPCRs, and DKK1 bind to CKAP4, PI3K becomes activated [49]. PI3K converts PIP2 (phosphatidylinositol 4,5-bisphosphate) into PIP3 (phosphatidylinositol 3,4,5-triphosphate), a key signaling molecule. PIP3 serves as a second messenger, recruiting AKT and PDK1 (3-phosphoinositide-dependent protein kinase 1) to the cell membrane, where AKT can be phosphorylated and fully activated. DKK1 significantly increases AKT phosphorylation in a dose-dependent manner, with AKT boosting the downstream signaling of DKK1 [50].
Analysis of DKK1 and active β-catenin expression in GC tissues confirmed concurrent DKK1 overexpression and abnormal activation of β-catenin signaling in GC compared to GIM and healthy mucosa, despite DKK1 being recognized as a canonical inhibitor of the Wnt pathway [12] (Figure 6).
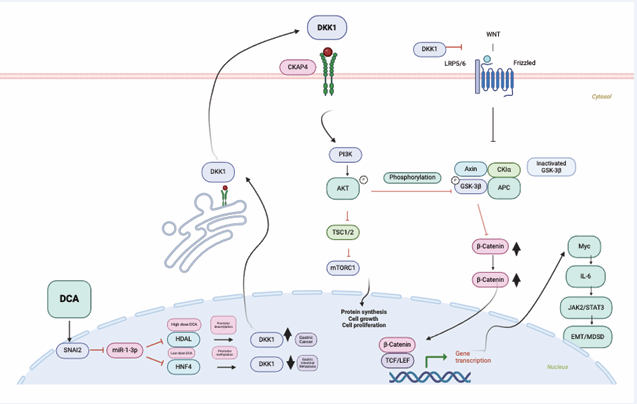
Figure 6: When DCA levels are low, HDAL6 and HNF4 cannot activate the DKK1 gene because of methylation at its promoter. However, when DCA levels increase enough, HDAL6 removes the repression of DKK1, allowing its levels to rise. DKK1 protein can be secreted into the blood, where it binds to its receptor CKAP4. This activates PI3K and phosphorylates AKT. P-AKT then inhibits GSK-3β, leading to increased cytoplasmic and nuclear β-catenin. Subsequently, β-catenin activates the Myc/IL-6/JAK2/STAT3 pathway.
Without Wnt signaling stimulation, β-catenin remains in a destruction complex composed of glycogen synthase kinase-3β (GSK-3β), casein kinase 1 (CK1), adenomatous polyposis coli (APC), and Axin. This complex promotes β-catenin phosphorylation, leading to its degradation via the ubiquitin-proteasome system [12]. The CKAP4 receptor activates the PI3K/AKT pathway and increases β-catenin levels by inhibiting phospho-GSK-3β. As a result, the phosphorylation and inhibition of GSK3β raise cytosolic β-catenin levels. Unphosphorylated β-catenin then translocates to the nucleus, where it interacts with T cell-specific factor (TCF)/lymphoid enhancer-binding factor (LEF) to activate target genes such as c-Myc, cyclin D1, CTNNB1, and DKK1. A strong positive correlation (P < 0.0001) was observed between DKK1 and CTNNB1 (the gene encoding β-catenin). C-Myc initiates transcription of downstream genes by binding to the Enhancer box (E-box) sequence and activating multiple signaling pathways.
The c-Myc proteins play a vital role in maintaining normal cellular function by regulating gene expression related to cell cycle progression, growth, and anti-apoptosis [12]. Mutations or overexpression of c-Myc genes significantly contribute to cancer development. The Myc gene is frequently overexpressed in various types of cancer, including GC [13].
The PDE5 inhibitor sildenafil has been shown to suppress oncogenic growth in GC. Sildenafil inhibits GC growth by directly activating PKG through PDE5 inhibition, which regulates c-MYC expression via its phosphorylation and ubiquitination, leading to decreased c-MYC stability. This results in the suppression of IL-6 transcription within the downstream IL-6/JAK/STAT3 signaling pathway [51]. LOC339059 inhibits M2 macrophage polarization, PD-L1 expression, tumor growth, and metastasis. It achieves this by reducing the activity of the IL-6/STAT3 signaling pathway and the transcriptional activation of IL-6, which is regulated by the c-Myc protein [52].
Unlimited Proliferation without Apoptosis: Cancer is a disease marked by an imbalance involving continuous cell growth and anti-apoptotic mechanisms. The β-catenin -TCF/LEF-dependent transcription machinery boosts the expression of MYC, SNAI1, PD-L1, and IL6/JAK2/ STAT3. These signaling pathways encourage the growth of rapidly dividing cancer stem cells (CSCs) and influence both immune surveillance and immune tolerance [44]. Several studies have explored how STAT3 promotes cell proliferation, survival, and anti-apoptotic pathways [53,54]. The primary ways STAT3 controls proliferation involve unchecked progression through the cell cycle. Cyclin D1 and c-Myc are involved in STAT3-driven abnormal cell cycle progression. STAT3 also enhances the expression of anti-apoptotic proteins, such as Bcl-2 and related family members, including BCL-XL and MCL-1, along with inhibitors of apoptosis like survivin and inhibitor of apoptosis protein-2 [28].
Evasion of immunity: Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) is a key immune checkpoint molecule, while the PD-1/PD-L1 pathway plays a vital role in regulating immune responses. Immunotherapy targeting PD-1/PD-L1 has shown promise in various cancers, as cancer cells expressing PD-L1 can exploit this pathway to evade immune attack. In human GC tissues, the expression of metastasis-associated colon cancer-1 (MACC-1), c-Met, and PD-L1 increased, and ectopic expression of MACC-1 led to higher levels of c-Met and PD-L1 [55]. Tumor-associated macrophages (TAMs) are crucial for establishing a suppressive tumor immune microenvironment (TIME) and promoting immune evasion, which significantly reduces the effectiveness of immune checkpoint inhibitors (ICIs) in GC. DKK1 causes macrophages to become immunosuppressive, impairing the antitumor responses of CD8+ T cells and NK cells. DKK1 interacts with CKAP4 on the macrophage surface and activates downstream PI3K-AKT signaling, contributing to immune suppression. Reprogramming TAMs by blocking DKK1 also improves the effectiveness of PD-1 blockade in GC [56].
DCA lowers the secretion of pro-inflammatory cytokines, including IL-6, IFN-γ, and TNF-α. It also impairs dendritic cell (DC) function, affecting their ability to release TNF-α and IL-12. Additionally, DCA diminishes NK cell activity by decreasing the secretion of IFN-γ and TNF-α. Chemokine ligand 28 (CCL28) is a direct transcriptional target of β-catenin/TCF. β-Catenin further promotes regulatory T cells, emphasizing the immunoregulatory role of β-catenin signaling in GC [44]. CNA analysis of early GC shows a gain of c-Myc, closely linked to the overactivation of the β-catenin signaling pathway. The positive rate of c-Myc expression is 100% for EGC, 94.4% for high-grade dysplasia, and 41.7% for low-grade dysplasia. In EGC, c-Myc expression is associated with nuclear and structural atypia [57]. Another study found that 77% of the GC exhibited Myc immunoreactivity [58].
During EMT, cancer cells acquire more invasive traits. The increased motility and invasiveness caused by EMT are essential for initiating metastasis and also improve resistance to chemotherapy. GC cells can secrete IL-6, which promotes tumor growth, development, and migration. Cancer-associated fibroblasts (CAFs) from GC produce large amounts of IL-6 [59]. Elevated levels of DCA in BR are linked to STAT3 activation during the transition from IM to GC. DCA stimulates the GPCR/STAT3/KLF5 pathway, encouraging cell proliferation and preventing apoptosis [13]. EGF impacts gene expression by affecting key signaling pathways, including Ras, PI3K, PLCγ, Myc, and STAT3. When STAT3 is activated, it highly promotes gastric carcinogenesis by regulating the expression of VEGF. CALM2 influences the JAK2/STAT3/HIF-1/VEGFA axis and supports the polarization of M2 macrophages [60].
DKK1 promotes an immunosuppressive TME (TIME) by activating downstream β-catenin/Myc/STAT3 signaling pathways. Tumor progression is linked to an increase in MDSCs, both within the TME and in the bloodstream. Myeloid-derived suppressor cells (MDSCs) accumulate at potential metastatic sites even before cancer cells start migrating [61]. A member of the Myc single-strand binding protein family, it helps regulate DNA replication, transcription, apoptosis, and cell cycle progression by interacting with the c-Myc protein. The key role of RBMS1 in promoting GC metastasis involves initiating autocrine IL-6/JAK2/STAT3 signaling. RBMS1 increases IL-6 expression and secretion through IL-6 transactivation by affecting histone modifications at the promoter regions after binding with the transcription factor Myc [53].
Phosphorylated STAT3 forms dimers and moves to the nucleus, where it activates the transcription of target genes, including those involved in immunosuppression, angiogenesis, metastasis, proliferation, and survival. The main mechanisms by which the immune system evades and tolerates tumors stem from dysfunctions in dendritic cells (DCs), MDSCs, TAMs, and tumor-associated neutrophils (TANs). T cells and natural killer (NK) cells become exhausted due to inhibitory signals such as cytokines (IL-6, IL-10) and growth factors (VEGF, TGF-β). Activation of immune checkpoint molecules like PD-1 and cytotoxic T-lymphocyte-associated (TIGIT) acts as negative regulators of the anti-tumor immune response [62].
DCs, which are antigen-presenting cells that initiate tumor-specific T-cell responses, have their activity suppressed by STAT3 activation. This activation blocks innate immune-stimulating molecules such as interferon-γ produced by CD8+ T cells, IL-12, TNF-α, and C-C Motif Chemokine Ligand 5 (CCL5), CCL9 [63]. Furthermore, it inhibits DC maturation by secreting IL-6, IL-10, and VEGF, which further activate STAT3 in a positive feedback loop. STAT3 activation is essential for the differentiation and expansion of MDSCs, which also help regulate innate immune responses by modulating cytokine production in macrophages. Macrophages can be classified as either classically activated M1 macrophages or alternatively activated M2 macrophages based on their interactions with cytokines produced by CD4+ T helper cell subpopulations [64]. M1 macrophages produce higher levels of pro inflammatory cytokines [65].
In contrast, M2 macrophages, which have elevated levels of immunosuppressive factors such as IL-10 and TGF-β, are considered pro-tumorigenic. Meanwhile, there is an inverse relationship with CD8+ T cells. This relationship reduces T cell functionality by modulating MDSCs and inhibiting the proliferation of CD8+ T cells and natural killer (NK) cells. MDSCs are widely recognized for their immunosuppressive effects and their ability to protect cancer cells from host immune responses [96]. These cells promote blood vessel formation and create metastatic niches. They are strongly associated with poor prognosis and resistance to ICI in the GC TIME. MDSCs stimulate arginase I, which helps suppress CD8+ T cells [66].
CD4+ T cells include CD4+ T-helper (Th) cells and regulatory T cells (Tregs), both of which play essential roles in cancer immunity. Furthermore, the peripheral blood of GC patients showed a higher frequency of IL-10 producing B cells compared to control individuals. Spatial profiling of primary tissue samples from GC patients confirmed that infiltrating MDSCs in cancerous tissues expressed markers typically linked to PMN-MDSCs, such as arginase-1 (ARG1), CD66B, VISTA, and indoleamine 2,3-dioxygenase 1 [78]. TANs are vital in tumor-related inflammation by promoting angiogenesis, metastasis, and immunosuppression [86,87]. Tumor-derived granulocyte macrophage colony-stimulating factor (GM-CSF) activates TANs and induces PD-L1 expression on these cells through JAK/STAT3 signaling [75]. MDSCs promote the expansion and induction of Treg cells by secreting IL-10 and IFN-γ. Additionally, MDSCs send signals that induce and develop Treg cells by upregulating ligands such as CD86, programmed death ligand 1 (PD-L1), and leukocyte immunoglobulin-like receptor subfamily B4 (LILRB4) [67].
Angiogenesis and Lymphangiogenesis: Angiogenesis is vital for the growth and spread of GC cells, as it supplies them with the necessary nutrients and oxygen. In tumor vessels, GPCR expression is markedly increased; blocking GPCR hampers endothelial cell (EC) migration during the formation of new blood vessels. GC cells produce more S1P, which is essential for EC migration and angiogenesis [68]. Hypoxic GC cells generate proangiogenic factors, including S1P and vascular endothelial growth factor (VEGF). The VEGFA-VEGFR2 signaling pathway plays a key role in promoting cancer neovascularization. Endothelial cell S1PR1 enhances VEGFA-VEGFR2-driven tumor angiogenesis. PVT1, a long non-coding RNA, supports angiogenesis and tumor growth by increasing nuclear STAT3 and stabilizing proteins [69]. Lymphangiogenesis in GC involves the Akt/mTOR-VEGF-C/VEGF-D axis, and the density of lymphatic vessels in GC tissues correlates with this pathway [70].
5.2.5. Immune Invasion and Metastasis: EMT is a process where epithelial cells lose their polarity and adhesion molecules while activating the cytoskeleton to become motile. The beginning of EMT involves reducing CDH1/E-cadherin expression. The abnormal activation of the EMT program is mainly driven by SNAI1/2, TWIST1/2, and ZEB1/2, as well as microRNAs. This ultimately increases genes associated with mesenchymal traits, such as vimentin, fibronectin, and N-cadherin [71]. Cancer cells undergo morphological changes that enable them to move actively through cytoskeletal reorganization. Gα12/13 plays a key role in cytoskeletal remodeling, cell migration, and invasion by activating the Rho/ROCK pathway. The β-catenin/Myc/STAT3 signaling pathway promotes migration and invasion in GC cells by activating focal adhesion kinase and Rac [72]. The ERK/MAPK signaling pathway promotes ECM breakdown through matrix metalloproteinases (MMPs), thereby enhancing tumor invasion and metastasis by increasing MMP expression. ADAM17 levels are significantly higher in GC and are associated with positive metastatic lymph nodes, showing a strong correlation with the survival times of GC patients. Molecules that interact with CasL (MICAL2) help facilitate E-cadherin ubiquitination and breakdown, leading to increased β-catenin signaling due to disruption of the E-cadherin/β-catenin complex, which then promotes GC migration [73].
Metastasis refers to how cancer cells spread to other organs or anatomical sites beyond the initial lesion and accounts for more than 90% of cancer-related deaths [74]. The most common sites for GC metastasis are the liver, lungs, bones, and lymph nodes. The steps in GC metastasis include invading surrounding tissue and degrading the basement membrane, entering blood vessels or the lymphatic system, surviving and moving to distant tissues, exiting into a new environment, and ultimately colonizing to grow and form a secondary tumor. The roles and mechanisms of GPCRs during invasion have been extensively studied [75].
CHEMOPREVENTION OF GC WITH VITAMIN D AND STATIN
Vitamin D (VTD)
VTD levels and GC: VTD deficiency increases both the incidence and mortality of GC. The risk of developing GC was approximately four times higher in cases with severe VTD (< 10 ng/mL) deficiency. VTD deficiency and VTD receptor polymorphism are significant risk factors for GC development [76]. Ten studies involving 1,159 GC patients and 33,387 controls showed that serum VTD levels in the GC group (15.56 ± 7.46 ng/mL) were lower than in the control group (17.60 ± 1.61 ng/mL), and this difference was statistically significant [80]. Research on the relationship between serum 25(OH)D levels and GC indicated that lower VTD levels increase the risk of GC. Results from nine case-control studies with 671 patients revealed that serum 25(OH)D levels in the GC group were lower than in controls (95% CI: −11.5, −6.32; p < 0.01). Additionally, the likelihood of VTD deficiency was higher in the GC group than in controls (odds ratio = 3.09, 95% CI: 1.96, 4.87; p < 0.01) [77]. VTD levels negatively correlated with GC and were significantly associated with clinical stages, degree of differentiation, and lymph node metastasis. This suggests that low VTD levels might be linked to a worse prognosis in GC [78]. The serum VTD level in the GC group (15.56 ± 7.46 ng/mL) was lower than in the control group (17.60 ± 1.61 ng/mL), with the difference being statistically significant. Patients with GC at clinical stages III/IV (16.19 ± 8.04 ng/ mL) had lower VTD levels compared to those at stages I/II (19.61 ± 9.61 ng/mL). Patients with poorly differentiated GC (17.5 ± 9.5 ng/mL) displayed lower levels than those with well- or moderately differentiated GC (18.04 ± 7.92 ng/mL). Furthermore, patients with lymph node metastasis (19.41 ± 8.63 ng/mL) had lower VTD levels than those without metastasis (20.65 ± 7.96 ng/mL) (p < 0.05) [79].
The prevalence of HP is highest in Africa compared to other regions of the world. Despite this high prevalence, GC rates in Africa remain low. This African perspective offers insights into the so-called “African Enigma” [80]. Studies conducted in India and several Asian countries, including Thailand, Bangladesh, Pakistan, Iran, Saudi Arabia, Israel, and Malaysia, report a high frequency of HP infection co-existing with a low incidence of GC [81]. The age-standardized rates of GC are highest in Northwest China, while South China has the lowest incidence rate [80]. Epidemiological data show that GC incidence and mortality are lower in sunny regions with high sunlight exposure compared to other areas. High UVB radiation correlates with a reduced incidence of GC (ES: 0.86, 95% CI: 0.84–0.89, P = 0) relative to regions with low UVB radiation [82].
Compared to normal tissues (82.61%) and premalignant tissues (73.64%), VDR expression was lower in GC tissues (57.61%) (p = 0.001). Among GC tissues, VDR expression was higher in well- and moderately differentiated tissues than in poorly differentiated ones. It was more elevated in smaller tumors (< 5 cm) than in larger tumors (≥ 5 cm) (p = 0.016, p = 0.009) [83]. Baseline concentrations of total (p<0.01), primary (p<0.01), and secondary (p<0.001) fecal BAs were significantly inversely related to baseline 25(OH)D levels. Individual bile acids, including CDCA (p<0.01), CA (p<0.05), LCA (p<0.001), DCA (p<0.001), and other bile acids (p<0.001), also showed significant inverse associations with 25(OH)D concentrations [84].
There is a continuous decline in GC risk associated with increased VTD intake in both men and women; fifth versus bottom quintile, OR, 95% CI: 0.68 (0.53, 0.86), OR, 95% CI: 0.72 (0.53, 0.97), OR, 95% CI: 0.58 (0.38, 0.89), respectively. Per increment quintile, the statistically significant reduction in GC risk was 7% in men and 13% in women [85].
Mechanism of vitamin D
VTDR to lower Cholesterol synthesis in the Liver: VTDR suppresses the hepatic small heterodimer partner (Shp) and Shp promoter activities that dominate over FXR mediated activation. It also increases levels of mouse and human CYP7A1 and decreases cholesterol synthesis in the liver [86]. VTD deficiency has been linked to a higher risk of CVD [87]. VTD deficiency was associated with significantly lower serum HDL-C (−5.1%) and higher total cholesterol (+9.4%), non-HDL-C (+15.4%), directly measured LDL-C (+13.5%), intermediate-density lipoprotein cholesterol (+23.7%), very low-density lipoprotein cholesterol (+19.0%), remnant lipoprotein cholesterol (+18.4%), and TG (+26.4%) compared to the optimal group [88].
VTD induced cell cycle arrest and Apoptosis: Vitamin D not only suppresses cancer cells and cancer stem cells but also regulates the tumor microenvironment, demonstrating the potential benefits of vitamin D in cancer prevention and treatment. 1,25(OH)2D3 has been shown to inhibit cell proliferation through cell cycle arrest in most cancer cells, which plays a vital role in cancer prevention. 1,25(OH)2D3 can induce p21 expression and G1 phase cell cycle arrest in a mutant p53 and VDR-dependent manner in gastric cancer cells. Meanwhile, p27 could be upregulated by 1,25(OH)2D3 [89]. VTDR is a member of the nuclear receptor family of transcription factors. The interaction between VTD and VTDR can trigger a sequence of gene regulation and cell signaling, which are crucial in anti-tumor processes, including inhibiting cell growth, promoting apoptosis and autophagy, blocking new blood vessel formation, and modulating the immune system [89].
1,25(OH)?D? influences GC development and growth by regulating multiple signaling pathways involved in cell proliferation, apoptosis, invasion, and metastasis [90,91]. 1,25(OH)?D? functions by binding to and activating the nuclear VTD receptor (VTDR), a ligand-modulated transcription factor that binds to specific sequences called VTD response elements (VDRE) in target genes. It also increases the expression of miR-145 and inhibits E2F3, CDK6, and other cell cycle regulatory genes like CDK2 and CCNA2, causing arrest at the G1 phase of the GC cell cycle. Additionally, this prevents the transition from G1 to S phase in the GC cell cycle.
The miR-145 target sequence of E2F3, a key gene involved in cell cycle regulation, is highly expressed in GC tissues. MiR-145 mediates the antiproliferative and gene regulatory effects of VTD in GC cells. Alongside miR-145, which targets multiple genes involved in the cell cycle, such as CDK6, c-Myc, and EGFR, E2F3 influences several genes implicated in cell cycle progression [92].
VTDR induces apoptosis. 1,25(OH)?D? enhances the transcription of several pro-apoptotic genes, including BAK, BAG, BIRC5, BAX, and G0S2, which encode proteins of the Bcl-2 family or interact with them to increase mitochondrial membrane permeability and trigger apoptosis [93]. 1,25(OH)?D? downregulates these proto oncogenes, boosts some of their functional antagonists, such as MAD/MXD1, and thereby counteracts uncontrolled cell growth. 1,25D inhibited MYC gene expression and accelerated its protein turnover [94].
VTD fights against β-catenin: The VTDR can significantly decrease the viability and invasive ability of gastric cancer cells. VTDR levels in GC cells treated with 1,25(OH)?D? showed a time-dependent increase in expression. As VDR expression rose, β-catenin levels gradually decreased, while E-cadherin levels showed a time-dependent increase (P < 0.05). When VTDR is activated by its ligand, it can prevent the nuclear import of β-catenin, influence E-cadherin levels, and inhibit the proliferation of gastric cancer cells, suggesting that the VTDR FokI gene may act as a cancer suppressor by blocking the β-catenin signaling pathway [89]. Reduced β-catenin signaling can prevent EMT occurrence, thereby inhibiting gastric cancer invasion and metastasis. EMT contributes to resistance against immunotherapy and anti-cancer agents. β-catenin accumulates in the nucleus and activates the transcription of downstream target genes such as c-myc, MMPs, and cyclin D1, which leads to abnormal cell proliferation in GC cells [95]. Its ligand activates VTDR, which can trigger a series of reactions to prevent β-catenin from entering the nucleus, thus inhibiting proliferation and regulating the invasion and migration of GC cells [89].
The overexpression of VTDR can significantly reduce the viability and invasive ability of GC cells. VTDR levels in GC cells treated with 1,25(OH)?D? show a time-dependent increase. As VDR levels rise, β-catenin levels gradually decrease, while E-cadherin levels increase over time (P < 0.05). When activated by its ligand, VTDR can block the nuclear import of β-catenin, affect E-cadherin levels, and inhibit GC cell proliferation [89]. Higher VTDR expression in GC cells results in lower β-catenin levels, significantly inhibiting cell growth. Conversely, lower VTDR levels lead to increased β-catenin, promoting cell proliferation, which further emphasizes VTDR’s role in regulating β-catenin to control GC cell proliferation and apoptosis [96].
VTD against Cancer Stem Cell: Cancer stem cells (CSCs) are a key factor in metastasis, recurrence, and chemotherapy resistance in cancer. It highlights targeting GC stem cells (GCSCs) as an effective treatment approach for GC [97]. CD44 is recognized as a marker for CSCs in various cancers, including GC. Both CD44 expression and the CD44-positive population are reduced by 1,25(OH)?D? through VTDR activation, both in vivo and in vitro, indicating that CD44 may mediate the action of VTD. VTD suppresses GC cells via the ß-catenin signaling pathway [45]. Vitamin D diminishes the stemness of cancer CSCs [98]. 1,25(OH)?D? induces the expression of E-cadherin and other adhesion proteins (occludin, Zonula occludens [ZO]-1, ZO-2, vinculin) and promotes the translocation of β-catenin and ZO-1 from the nucleus to the plasma membrane. Ligand-activated VDR competes with the T cell transcription factor (TCF)-4 for β-catenin binding. Accordingly, 1,25(OH)?D? represses β-catenin–TCF-4 transcriptional activity. Additionally, 1,25(OH)?D? inhibits the expression of β–catenin–TCF4–responsive genes, including c-myc, PPARγ, Tcf-1, and CD44, while inducing the expression of ZO-1 [99] (Figure 7).
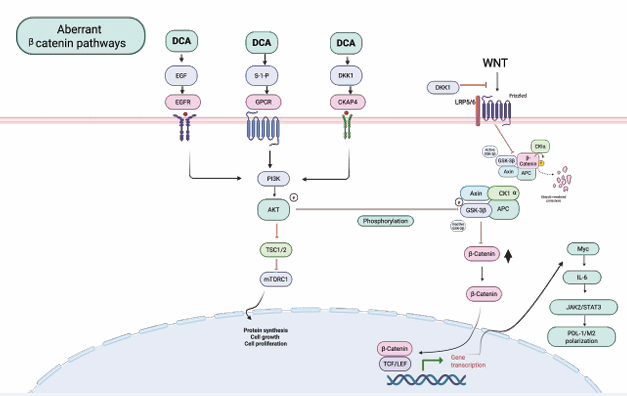
Figure 7: EGF, GPCRs, and DKK1, which DCA activates, bind to CKAP4. This binding triggers the activation of PI3K, which then phosphorylates AKT. P-AKT inhibits GSK-3β, resulting in increased levels of both cytoplasmic and nuclear β-catenin. Consequently, β-catenin activates the Myc/ IL-6/JAK2/STAT3 pathway. DKK1 raises vascular endothelial growth factor levels and also promotes tumor immune suppression, immune evasion, chemoresistance, and metastasis.
VTD and Gut Microbiome: VTD reduces the permeability of intestinal cells in animal models of colitis. In VTDR knock-out mice, dextran sodium sulfate induces colitis that is associated with decreased immunostaining of zonula occludens-1 and occludin proteins on colon epithelial cells, along with lower transepithelial resistance and increased permeability. The nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is an intracellular pattern recognition receptor. It activates NF-kB and boosts VTD-mediated transcription of cathelicidin and DEFB4 (defensin, beta 4) [100]. Mice with higher levels of VTD show increased immune-dependent resistance to transplantable cancers and stronger responses to checkpoint blockade immunotherapies. In mice, this resistance results from VTD’s activity on intestinal epithelial cells, which changes the microbiome composition and promotes Bacteroides fragilis, thereby boosting cancer immunity. Disruption of VTD signaling in intestinal epithelial cells (IECs) alters the microbiome and subsequently affects cancer immunity in mice. In humans, VTD-induced genes are linked with better responses to immune checkpoint inhibitor treatment, improved cancer immunity, and increased survival [101].
Intestinal VTDR conditional knockout (VTDRΔIEC) mice show an increased rate of tumor formation. Fecal microbiota analysis demonstrated that VTDR deletion causes a shift in bacterial profiles toward those more linked to carcinogenesis. Microbial metabolites from VTDRΔIEC mice displayed higher levels of DCA and LCA. The JAK/STAT pathway is essential for maintaining intestinal and microbial homeostasis. Fecal samples from VTDRΔIEC mice activate STAT3 signaling in human and mouse organoids [102].
VTD levels appear to regulate the abundance and/or metabolic functions of Bacteroides fragilis, an anaerobic Gram-negative bacterium that is part of the normal microbiome in humans and mice. Increased vitamin D availability improves immune-dependent resistance to transplantable cancers and enhances responses to checkpoint blockade immunotherapies. Furthermore, VTD supplementation in healthy human volunteers is associated with a significant increase in intestinal Bacteroides species and the Bacteroides/Prevotella ratio. In both humans and mice, VTD activity seems to strengthen immune responses to cancer [103]. Human VTD levels influence how intestinal cells produce mediators that promote a modified microbiome, including organisms like Bacteroides fragilis, which can boost cancer immunity. VTD can inhibit cancer cell growth, promote apoptosis, and reduce angiogenesis. Additionally, the VTD-VDR gene signature is associated with markers of anti-cancer immunity and patient responses to immunotherapy [103].
Low-Density Lipoprotein and Statins
Studies have repeatedly shown that, regardless of physical activity levels and nutritional status, low-density lipoprotein (LDL) cholesterol levels are widely accepted to increase with age in both sexes across various groups [104,105]. LDL is a critical lipoprotein that carries cholesterol, mediating the transfer of cholesterol from the liver to peripheral tissues. It has been observed that the LDL receptor (LDLR) is overexpressed in various types of cancer. The LDLRs represent a family of pleiotropic cell surface receptors involved in lipid homeostasis, cell migration, proliferation, and differentiation [106]. Abnormal lipid metabolism can lead to lipotoxicity, triggering oxidative stress and significantly increasing the levels of ROS. A gradual buildup of oxidative stress can cause LDL to become oxidized, resulting in the formation of ox-LDL. Additionally, oxidative stress promotes DNA damage in cancers, contributing to malignant transformation and carcinogenesis. Elevated plasma oxLDL has been observed in GC [107]. Plasma oxLDL was positively correlated with lymphatic metastasis in patients with GC. Additionally, oxLDL promoted the expression and secretion of VEGF in GC cell lines [108].
Reflux of BAs from the duodenum into the stomach can harm the gastric epithelium, leading to IM and a higher risk of GC. Statins, cholesterol-lowering drugs, decrease the amount of BAs in the liver, thereby lowering BR to the stomach. In a systematic review and meta-analysis examining the association between statin use and GC, the group using statins demonstrated a significantly lower risk of GC compared to the non-statin group (OR/RR, 0.74; 95% CI: 0.67-0.80, P < 0.001). Additionally, the statin group showed significantly lower all-cause mortality and GC-specific mortality than the no-statin group (all-cause mortality: HR, 0.70; 95% CI: 0.52-0.95, P = 0.021; GC-specific mortality: HR, 0.70; 95% CI: 0.58-0.84, P < 0.001) [109]. Another meta-analysis found that statin use was associated with a reduced risk of GC (relative risk: 0.72; 95% CI: 0.64-0.81, p < 0.001) [110]. In Taiwan, statin use improved the overall survival of patients with GC after surgery and adjuvant chemotherapy (hazard ratio 0.62; 95% CI: 0.50-0.78) [111]. Statin use also reduced-cancer specific mortality in GC patients in the UK (adjusted HR, 0.83; 95% CI, 0.73-0.94) [110]. The use of statins is associated with a significantly lower risk of developing GC in several population-based cohort studies [111-114].
Low VTD levels are linked to hypercholesterolemia, and VTD supplements can reduce total and LDL cholesterol [115]. VTD inhibits hepatic Cyp71α1 through FXR in the liver via small heterodimer protein (SHP) and in the terminal ileum via intestinal fibroblast growth factor (FGF) 15/19 [32]. VTD protects against atherosclerosis by regulating cholesterol efflux and macrophage polarization in hypercholesterolemic swine. 1,25(OH)2D3 significantly increases Cyp27A1 expression via a VTDR-dependent JNK1/2 signaling pathway. It raises 27-hydroxycholesterol levels, which induce LXRs, ABCA1, and ABCG1 expression, stimulating cholesterol efflux that VTDR antagonists and JNK1/2 signaling inhibitors can block in THP-1 macrophage-derived foam cells [116]. Statins have anticancer effects by inhibiting the proliferation, cell cycle progression, migration, and invasion of GC cells through the suppression of Interleukin Enhancing Factor 3 (ILF3) expression [95]. They work synergistically with PD-1 inhibitors to improve the prognosis for patients with GC. Statins effectively decrease serum levels of PD-L1 and interleukin-enhancer binding factor 3 (ILF3), thereby boosting patient outcomes. Specifically, simvastatin promotes the overexpression of HDAC6 and reduces acetylation at the H3K14 residue in ILF3, leading to lower ILF3 expression. Lowering ILF3 induces ferroptosis in GC cells by regulating SLC7A11/GPX4 through the PI3K/ AKT/mTOR signaling pathway [117]. Additionally, ILF3 helps recruit activated CD8+ T cells by reducing PD-L1 expression, which enhances their ability to kill GC cells [118].
CONCLUSION AND FUTURE DIRECTIONS
Hydrophobic BAs from duodenogastric reflux can cause GIM, which is linked to AG. CDCA, a hydrophobic primary BA, stimulates FXR in the gastric epithelium and triggers CIM. DCA, one of the most cytotoxic BAs, induces IIM and GC by disrupting mitochondrial outer membranes and producing reactive oxygen and nitrogen species that damage DNA and chromosomes. DCA promotes gastric carcinogenesis by activating GPCR, EGFR, β-catenin, and DKK1, leading to increased cell proliferation, migration, invasion, metastasis, angiogenesis, and immune evasion. Lowering DCA levels in refluxed bile could help reduce GC incidence and mortality. VTD supplements might be an affordable, well-tolerated anti-cancer option against GC. VTD deficiency is a key factor in developing gastric cancer. VTD exhibits anti-tumor effects by inducing apoptosis and inhibiting β-catenin signaling pathways in gastric cancer. Numerous studies show that high serum levels of 25(OH) vitamin D3 are protective against GC. Statins reduce cholesterol synthesis and duodenogastric bile reflux. A randomized, double-blind, controlled study should be the primary focus of future research to demonstrate the actual anticancer effect of VTD against GC when combined with other anti-cancer agents.
REFERENCES
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021; 71: 209-249.
- de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health. 2020; 8: e180-e190.
- Yasuda T, Wang YA. Gastric cancer immunosuppressive microenvironment heterogeneity: implications for therapy development. Trends Cancer. 2024; 10: 627-642.
- Chen HN, Wang Z, Li X, Zhou ZG. Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: evidence from a meta-analysis. Gastric Cancer. 2016; 19: 166-175.
- Cheng Y, Wang S, Zhu W, Xu Z, Xiao L, Wu J, et al. Deoxycholic acid inducing chronic atrophic gastritis with colonic mucosal lesion correlated to mucosal immune dysfunction in rats. Sci Rep. 2024; 14: 15798.
- Tatsugami M, Ito M, Tanaka S, Yoshihara M, Matsui H, Haruma K, et al. Bile acid promotes intestinal metaplasia and gastric carcinogenesis. Cancer Epidemiol Biomarkers Prev. 2012; 21: 2101-2107.
- He Q, Liu L, Wei J, Jiang J, Rong Z, Chen X, et al. Roles and action mechanisms of bile acid-induced gastric intestinal metaplasia: a review. Cell Death Discov. 2022; 8: 158.
- Angelin B, Björkhem I, Einarsson K, Ewerth S. Hepatic uptake of bile acids in man. Fasting and postprandial concentrations of individual bile acids in portal venous and systemic blood serum. J Clin Invest. 1982; 70: 724-731.
- Qu X, Shi Y. Bile reflux and bile acids in the progression of gastric intestinal metaplasia. Chin Med J (Engl). 2022; 135: 1664-1672.
- Kouhzad M, Götz F, Navidifar T, Taki E, Ghamari M, Mohammadzadeh R, et al. Carcinogenic and anticancer activities of microbiota-derived secondary bile acids. Front Oncol. 2025; 15: 1514872.
- Xiang J, Zhang Z, Xie H, Zhang C, Bai Y, Cao H, et al. Effect of different bile acids on the intestine through enterohepatic circulation based on FXR. Gut Microbes. 2021; 13: 1949095.
- Shah SC, Gawron AJ, Mustafa RA, Piazuelo MB. Histologic Subtyping of Gastric Intestinal Metaplasia: Overview and Considerations for Clinical Practice. Gastroenterology. 2020; 158: 745-750.
- Jin D, Huang K, Xu M, Hua H, Ye F, Yan J, et al. Deoxycholic acid induces gastric intestinal metaplasia by activating STAT3 signaling and disturbing gastric bile acids metabolism and microbiota. Gut Microbes. 2022; 14: 2120744.
- Li J, Zhang Y, Ye F, Qian P, Qin Z, Li D, et al. DKK1 Promotes Epithelial- Mesenchymal Transition and Cisplatin Resistance in Gastric Cancer via Activation of the PI3K/AKT Pathway. Cancers (Basel). 2023; 15: 4756.
- Du S, Yang Y, Fang S, Guo S, Xu C, Zhang P, et al. Gastric Cancer Risk of Intestinal Metaplasia Subtypes: A Systematic Review and Meta- Analysis of Cohort Studies. Clin Transl Gastroenterol. 2021; 12: e00402.
- Li D, Zhang J, Yao WZ, Zhang DL, Feng CC, He Q, et al. The relationship between gastric cancer, its precancerous lesions and bile reflux: A retrospective study. J Dig Dis. 2020; 21: 222-229.
- Yu JH, Zheng JB, Qi J, Yang K, Wu YH, Wang K, et al. Bile acids promote gastric intestinal metaplasia by upregulating CDX2 and MUC2 expression via the FXR/NF-κB signalling pathway. Int J Oncol. 2019; 54: 879-892.
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999; 284: 1362-1365.
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003; 278: 9435-9440.
- Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones.Steroids. 2014; 86: 62-68.
- Yao Y, Li X, Xu B, Luo L, Guo Q, Wang X, et al. Cholecystectomy promotes colon carcinogenesis by activating the Wnt signaling pathway by increasing the deoxycholic acid level. Cell Commun Signal. 2022; 20: 71.
- Lu W, Ni Z, Tong M, Jiang S, Zhang J, Feng C, et al. DKK1 is epigenetically downregulated by promoter methylation and inhibits bile acid-induced gastric intestinal metaplasia. Biochem Biophys Res Commun. 2020; 523: 780-786.
- Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006; 47: 241-259.
- Wang N, Chen M, Ni Z, Li T, Zeng J, Lu G, et al. HDAC6/HNF4α loop mediated by miR-1 promotes bile acids-induced gastric intestinal metaplasia. Gastric Cancer. 2021; 24: 103-116.
- Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol. 2011; 85: 863-871.
- Centuori SM, Gomes CJ, Trujillo J, Borg J, Brownlee J, Putnam CW, et al. Deoxycholic acid mediates non-canonical EGFR-MAPK activation through the induction of calcium signaling in colon cancer cells. Biochim Biophys Acta. 2016; 1861: 663-670.
- Jenkins GJ, D’Souza FR, Suzen SH, Eltahir ZS, James SA, Parry JM, et al. Deoxycholic acid at neutral and acid pH, is genotoxic to oesophageal cells through the induction of ROS: The potential role of anti-oxidants in Barrett’s oesophagus. Carcinogenesis. 2007; 28: 136-142.
- Hu Y, Dong Z, Liu K. Unraveling the complexity of STAT3 in cancer: molecular understanding and drug discovery. J Exp Clin Cancer Res. 2024; 43: 23.
- Liu Y, Zhang S, Zhou W, Hu D, Xu H, Ji G. Secondary Bile Acids and Tumorigenesis in Colorectal Cancer. Front Oncol. 2022; 12: 813745.
- Prigent C. Centriole Duplication at the Crossroads of Cell Cycle Control and Oncogenesis. Cells. 2025; 14: 1094.
- Lei ZN, Teng QX, Tian Q, Chen W, Xie Y, Wu K, et al. Signaling pathways and therapeutic interventions in gastric cancer. Signal Transduct Target Ther. 2022; 7: 358.
- Yan H, Zhang JL, Leung KT, Lo KW, Yu J, To KF, et al. An Update of G-Protein-Coupled Receptor Signaling and Its Deregulation in Gastric Carcinogenesis. Cancers (Basel). 2023; 15: 736.
- Dong W, Liu L, Dou Y, Xu M, Liu T, Wang S, et al. Deoxycholic acid activates epidermal growth factor receptor and promotes intestinal carcinogenesis by ADAM17-dependent ligand release. J Cell Mol Med. 2018; 22: 4263-4273.
- Dong W, Liu L, Dou Y, Xu M, Liu T, Wang S, et al. Deoxycholic acid activates epidermal growth factor receptor and promotes intestinal carcinogenesis by ADAM17-dependent ligand release. J Cell Mol Med. 2018; 22: 4263-4273.
- Ding Y, Li H, Cao S, Yu Y. Effects of catechin on the malignant biological behavior of gastric cancer cells through the PI3K/Akt signaling pathway. Toxicol Appl Pharmacol. 2024; 490: 117036.
- Zhang Y, Wang Y, Wan Z, Liu S, Cao Y, Zeng Z. Sphingosine kinase 1 and cancer: a systematic review and meta-analysis. PLoS One. 2014; 9: e90362.
- He Y, Ge Y, Jiang M, Zhou J, Luo D, Fan H, et al. MiR-592 Promotes Gastric Cancer Proliferation, Migration, and Invasion Through the PI3K/AKT and MAPK/ERK Signaling Pathways by Targeting Spry2. Cell Physiol Biochem. 2018; 47: 1465-1481.
- You X, Wang Y, Wu J, Liu Q, Chen D, Tang D, et al. Galectin-1 Promotes Metastasis in Gastric Cancer Through a Sphingosine-1-Phosphate Receptor 1-Dependent Mechanism. Cell Physiol Biochem. 2018; 51: 11-30.
- Ma Z, Sun Q, Zhang C, Zheng Q, Liu Y, Xu H, et al. RHOJ Induces Epithelial-to-Mesenchymal Transition by IL-6/STAT3 to Promote Invasion and Metastasis in Gastric Cancer. Int J Biol Sci. 2023; 19: 4411-4426.
- Liu AM, Wong YH. Activation of nuclear factor {kappa}B by somatostatin type 2 receptor in pancreatic acinar AR42J cells involves G{alpha}14 and multiple signaling components: a mechanism requiring protein kinase C, calmodulin-dependent kinase II, ERK, and c-Src. J Biol Chem. 2005; 280: 34617-34625.
- Shida D, Kitayama J, Yamaguchi H, Yamashita H, Mori K, Watanabe T, et al. Sphingosine 1-phosphate transactivates c-Met as well as epidermal growth factor receptor (EGFR) in human gastric cancer cells. FEBS Lett. 2004; 577: 333-338.
- Yasuda H, Hirata S, Inoue K, Mashima H, Ohnishi H, Yoshiba M. Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem Biophys Res Commun. 2007; 354: 154-159.
- Morgos DT, Stefani C, Miricescu D, Greabu M, Stanciu S, Nica S, et al. Targeting PI3K/AKT/mTOR and MAPK Signaling Pathways in Gastric Cancer. Int J Mol Sci. 2024; 25: 1848.
- Katoh M, Katoh M. WNT signaling and cancer stemness. Essays Biochem. 2022; 66: 319-331.
- Li Q, Li Y, Jiang H, Xiao Z, Wu X, Zhang H, et al. Vitamin D suppressed gastric cancer cell growth through downregulating CD44 expression in vitro and in vivo. Nutrition. 2021; 91-92: 111413.
- Zhuang GF, Tan Y, Zeng JT, Zhang JW, Tang J, Zeng SP, et al. Expression of serum Dickkopf-1 in gastric cancer patients. Asian Pac J Trop Med. 2015; 8: 870-872.
- Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, et al. DKK1, a negative regulator of Wnt signaling, is a target of the beta- catenin/TCF pathway. Oncogene. 2004; 23: 8520-8526.
- Kimura H, Yamamoto H, Harada T, Fumoto K, Osugi Y, Sada R, et al. CKAP4, a DKK1 Receptor, Is a Biomarker in Exosomes Derived from Pancreatic Cancer and a Molecular Target for Therapy. Clin Cancer Res. 2019; 25: 1936-1947.
- Katoh M, Katoh M. Molecular genetics and targeted therapy of WNT- related human diseases (Review). Int J Mol Med. 2017; 40: 587-606.
- Li J, Zhang Y, Ye F, Qian P, Qin Z, Li D, et al. DKK1 Promotes Epithelial- Mesenchymal Transition and Cisplatin Resistance in Gastric Cancer via Activation of the PI3K/AKT Pathway. Cancers (Basel). 2023; 15: 4756.
- Riihimäki M, Hemminki A, Sundquist K, Sundquist J, Hemminki K. Metastatic spread in patients with gastric cancer. Oncotarget. 2016; 7: 52307-52316.
- Durak ?, Gheybi A, Demirkol ?, Ar?kan S, Zeybek ?Ü, Akyüz F, et al. The effects of serum levels, and alterations in the genes of binding protein and receptor of vitamin D on gastric cancer. Mol Biol Rep. 2019; 46: 6413-6420.
- Liu M, Li H, Zhang H, Zhou H, Jiao T, Feng M, et al. RBMS1 promotes gastric cancer metastasis through autocrine IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2022; 13: 287.
- Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in Cancer Immunotherapy. Mol Cancer. 2020; 19: 145.
- Tong G, Cheng B, Li J, Wu X, Nong Q, He L, et al. MACC1 regulates PDL1 expression and tumor immunity through the c-Met/AKT/mTOR pathway in gastric cancer cells. Cancer Med. 2019; 8: 7044-7054.
- Haas MS, Kagey MH, Heath H, Schuerpf F, Rottman JB, Newman W. mDKN-01, a Novel Anti-DKK1 mAb, Enhances Innate Immune Responses in the Tumor Microenvironment. Mol Cancer Res. 2021; 19: 717-725.
- Arakawa N, Irisawa A, Ishida K, Tsunoda T, Yamaguchi Y, Shibukawa G, et al. Clinical Differences in c-Myc Expression in Early-Stage Gastric Neoplasia: A Retrospective Study Based on the WHO Classification. J Clin Med. 2022; 11: 544.
- de Souza CR, Leal MF, Calcagno DQ, Costa Sozinho EK, Borges Bdo N, Montenegro RC, et al. MYC deregulation in gastric cancer and its clinicopathological implications. PLoS One. 2013; 8: e64420.
- Wei R, Song J, Pan H, Liu X, Gao J. CPT1C-positive cancer-associated fibroblast facilitates immunosuppression through promoting IL-6- induced M2-like phenotype of macrophage. Oncoimmunology. 2024; 13: 2352179.
- Mu G, Zhu Y, Dong Z, Shi L, Deng Y, Li H. Calmodulin 2 Facilitates Angiogenesis and Metastasis of Gastric Cancer via STAT3/HIF-1A/ VEGF-A Mediated Macrophage Polarization. Front Oncol. 2021; 11: 727306.
- Shi T, Zhang Y, Wang Y, Song X, Wang H, Zhou X, et al. DKK1 Promotes Tumor Immune Evasion and Impedes Anti-PD-1 Treatment by Inducing Immunosuppressive Macrophages in Gastric Cancer. Cancer Immunol Res. 2022; 10: 1506-1524.
- Tang W, Pan X, Han D, Rong D, Zhang M, Yang L, et al. Clinical significance of CD8+ T cell immunoreceptor with Ig and ITIM domains+ in locally advanced gastric cancer treated with SOX regimen after D2 gastrectomy. Oncoimmunology. 2019; 8: e1593807.
- Katayama N, Ohuchida K, Son K, Tsutsumi C, Mochida Y, Noguchi S, et al. Tumor infiltration of inactive CD8 + T cells was associated with poor prognosis in Gastric Cancer. Gastric Cancer. 2025; 28: 211-227.
- Oya Y, Hayakawa Y, Koike K. Tumor microenvironment in gastric cancers. Cancer Sci. 2020; 111: 2696-2707.
- Ma J, Shi Y, Lu Q, Huang D. Inflammation-Related Gene ADH1A Regulates the Polarization of Macrophage M1 and Influences the Malignant Progression of Gastric Cancer. J Inflamm Res. 2024; 17: 4647-4665.
- Deng C, Huo M, Chu H, Zhuang X, Deng G, Li W, et al. Exosome circATP8A1 induces macrophage M2 polarization by regulating the miR-1-3p/STAT6 axis to promote gastric cancer progression. Mol Cancer. 2024. 23.
- Su MT, Kumata S, Endo S, Okada Y, Takai T. LILRB4 promotes tumor metastasis by regulating MDSCs and inhibiting miR-1 family miRNAs. Oncoimmunology. 2022; 11: 2060907.
- Xu G, Yang Z, Sun Y, Dong H, Ma J. Interaction of microRNAs with sphingosine kinases, sphingosine-1 phosphate, and sphingosine-1 phosphate receptors in cancer. Discov Oncol. 2021; 12: 33.
- Zhao J, Du P, Cui P, Qin Y, Hu C, Wu J, et al. LncRNA PVT1 promotes angiogenesis via activating the STAT3/VEGFA axis in gastric cancer. Oncogene. 2018 Jul; 37: 4094-4109.
- Chen H, Guan R, Lei Y, Chen J, Ge Q, Zhang X, et al. Lymphangiogenesis in gastric cancer regulated through Akt/mTOR-VEGF-C/VEGF-D axis. BMC Cancer. 2015; 15: 103.
- Xu C, Li M, Zhang L, Bi Y, Wang P, Li J, et al. MicroRNA-205 suppresses the invasion and epithelial-mesenchymal transition of human gastric cancer cells. Mol Med Rep. 2016; 13: 4767-4773.
- Wang W, Wang M, Du T, Hou Z, You S, Zhang S, et al. SHMT2 Promotes Gastric Cancer Development through Regulation of HIF1α/VEGF/ STAT3 Signaling. Int J Mol Sci. 2023; 24: 7150.
- Wang Q, Qi C, Min P, Wang Y, Ye F, Xia T, et al. MICAL2 contributes to gastric cancer cell migration via Cdc42-dependent activation of E-cadherin/β-catenin signaling pathway. Cell Commun Signal. 2022; 20: 136.
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006; 12: 895-904.
- Katoh M, Katoh M. Molecular genetics and targeted therapy of WNT- related human diseases (Review). Int J Mol Med. 2017; 40: 587-606.
- Zhang Y, Li Y, Wei Y, Cong L. Molecular Mechanism of Vitamin D Receptor Modulating Wnt/β-catenin Signaling Pathway in Gastric Cancer. J Cancer. 2023; 14: 3285-3294.
- Liu X, Zhou Y, Zou X. Correlation between Serum 25-Hydroxyvitamin D Levels and Gastric Cancer: A Systematic Review and Meta-Analysis. Curr Oncol. 2022; 29: 8390-8400.
- Ren C, Qiu MZ, Wang DS, Luo HY, Zhang DS, Wang ZQ, et al. Prognostic effects of 25-hydroxyvitamin D levels in gastric cancer. J Transl Med. 2012; 10: 16.
- Zhao X, Wang J, Zou L. Vitamin D and gastric cancer - A systematic review and meta-analysis. Nutr Hosp. 2023; 40: 1080-1087.
- Holcombe C. Helicobacter pylori: the African enigma. Gut. 1992; 33: 429-431.
- Graham DY, Lu H, Yamaoka Y. African, Asian or Indian enigma, the East Asian Helicobacter pylori: facts or medical myths. J Dig Dis. 2009; 10: 77-84.
- Chen X, Li L, Liang Y, Huang T, Zhang H, Fan S, Sun W, Wang Y. Relationship of vitamin D intake, serum 25(OH) D, and solar ultraviolet-B radiation with the risk of gastric cancer: A meta- analysis. J Cancer Res Ther. 2022; 18: 1417-1424.
- Wen Y, Da M, Zhang Y, Peng L, Yao J, Duan Y. Alterations in vitamin D signaling pathway in gastric cancer progression: a study of vitamin D receptor expression in human normal, premalignant, and malignant gastric tissue. Int J Clin Exp Pathol. 2015; 8: 13176-13184.
- Khayatzadeh S, Feizi A, Saneei P, Esmaillzadeh A. Vitamin D intake, serum Vitamin D levels, and risk of gastric cancer: A systematic review and meta-analysis. J Res Med Sci. 2015; 20: 790-796.
- Nguyen MT, Huynh NNY, Nguyen DD, Ta NH, Van Nguyen T, Dang HT, et al. Vitamin D intake and gastric cancer in Viet Nam: a case-control study. BMC Cancer. 2022; 22: 838.
- Chow EC, Magomedova L, Quach HP, Patel R, Durk MR, Fan J, et al. Vitamin D receptor activation down-regulates the small heterodimer partner and increases CYP7A1 to lower cholesterol. Gastroenterology. 2014; 146: 1048-1059.
- Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med. 2008; 168: 1174-1180.
- Dibaba DT. Effect of vitamin D supplementation on serum lipid profiles: a systematic review and meta-analysis. Nutr Rev. 2019; 77: 890-902.
- Zhang Y, Li Y, Wei Y, Cong L. Molecular Mechanism of Vitamin D Receptor Modulating Wnt/β-catenin Signaling Pathway in Gastric Cancer. J Cancer. 2023; 14: 3285-3294.
- Li M, Li L, Zhang L, Hu W, Shen J, Xiao Z, et al. 1,25-Dihydroxyvitamin D3 suppresses gastric cancer cell growth through VDR- and mutant p53-mediated induction of p21. Life Sci. 2017; 179: 88-97.
- Bao A, Li Y, Tong Y, Zheng H, Wu W, Wei C. 1,25-Dihydroxyvitamin D? and cisplatin synergistically induce apoptosis and cell cycle arrest ingastric cancer cells. Int J Mol Med. 2014; 33: 1177-1184.
- Chang S, Gao L, Yang Y, Tong D, Guo B, Liu L, et al. miR-145 mediates the antiproliferative and gene regulatory effects of vitamin D3 by directly targeting E2F3 in gastric cancer cells. Oncotarget. 2015; 6: 7675-7685.
- Guzey M, Kitada S, Reed JC. Apoptosis induction by 1alpha, 25-dihydroxyvitamin D3 in prostate cancer. Mol Cancer Ther. 2002; 1: 667-677.
- Salehi-Tabar R, Nguyen-Yamamoto L, Tavera-Mendoza LE, Quail T, Dimitrov V, An BS, et al. Vitamin D receptor as a master regulator of the c-MYC/MXD1 network. Proc Natl Acad Sci U S A. 2012; 109: 18827-18832.
- Yu J, Sun Q, Hui Y, Xu J, Shi P, Chen Y, et al. Vitamin D receptor prevents tumour development by regulating the Wnt/β-catenin signalling pathway in human colorectal cancer. BMC Cancer. 2023; 23: 336.
- Xu S, Zhang ZH, Fu L, Song J, Xie DD, Yu DX, et al. Calcitriol inhibits migration and invasion of renal cell carcinoma cells by suppressing Smad2/3-, STAT3- and β-catenin-mediated epithelial-mesenchymal transition. Cancer Sci. 2020; 111: 59-71.
- Rao X, Zhang C, Luo H, Zhang J, Zhuang Z, Liang Z, et al. Targeting Gastric Cancer Stem Cells to Enhance Treatment Response. Cells. 2022; 11: 2828.
- Ji M, Liu L, Hou Y, Li B. 1α,25-Dihydroxyvitamin D3 restrains stem cell-like properties of ovarian cancer cells by enhancing vitamin D receptor and suppressing CD44. Oncol Rep. 2019; 41: 3393-3403.
- Pálmer HG, González-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001; 154: 369-387.
- Kamr AM, Bartish C, Summers J, Horton J, Hostnik LD, Orr K, et al. Longitudinal Evaluation of Vitamin D, Parathyroid Hormone, Antimicrobial Peptides, and Immunomodulatory Genes in Hospitalized Foals. J Vet Intern Med. 2025; 39: e70012.
- Sun J, Zhang YG. Vitamin D Receptor Influences Intestinal Barriers in Health and Disease. Cells. 2022; 11: 1129.
- Zhang YG, Lu R, Wu S, Chatterjee I, Zhou D, Xia Y, et al. Vitamin D Receptor Protects Against Dysbiosis and Tumorigenesis via the JAK/ STAT Pathway in Intestine. Cell Mol Gastroenterol Hepatol. 2020; 10: 729-746.
- Giampazolias E, Pereira da Costa M, Lam KC, Lim KHJ, Cardoso A, Piot C, et al. Vitamin D regulates microbiome-dependent cancer immunity. Science. 2024; 384: 428-437.
- Mc Auley MT, Wilkinson DJ, Jones JJ, Kirkwood TB. A whole- body mathematical model of cholesterol metabolism and its age- associated dysregulation. BMC Syst Biol. 2012; 6: 130.
- Hayashi R, Kogure S, Sakaibori K, Kurihara S, Kasagi J, Murata K. Serum cholesterol level in normal people: association of serum cholesterol level with age and relative body weight. Jpn J Med. 1987; 26: 153-157.
- Passarella D, Ciampi S, Di Liberto V, Zuccarini M, Ronci M, Medoro A, et al. Low-Density Lipoprotein Receptor-Related Protein 8 at the Crossroad between Cancer and Neurodegeneration. Int J Mol Sci. 2022; 23: 8921.
- Deng CF, Zhu N, Zhao TJ, Li HF, Gu J, Liao DF, et al. Involvement of LDL and ox-LDL in Cancer Development and Its Therapeutical Potential. Front Oncol. 2022; 12: 803473.
- Ma C, Xie J, Luo C, Yin H, Li R, Wang X, et al. OxLDL promotes lymphangiogenesis and lymphatic metastasis in gastric cancer by upregulating VEGF-C expression and secretion. Int J Oncol. 2019; 54: 572-584.
- Chen Y, Zhang J, Zhang Y, Zhu L. Effect of statin use on risk and mortality of gastric cancer: a meta-analysis. Anticancer Drugs. 2023; 34: 901-909.
- Spence AD, Busby J, Hughes CM, Johnston BT, Coleman HG, Cardwell CR. Statin use and survival in patients with gastric cancer in two independent population-based cohorts. Pharmacoepidemiol Drug Saf. 2019; 28: 460-470.
- Yang PR, Tsai YY, Chen KJ, Yang YH, Shih WT. Statin Use Improves Overall Survival of Patients with Gastric Cancer after Surgery and Adjuvant Chemotherapy in Taiwan: A Nationwide Matched Cohort Study. Cancers (Basel). 2020; 12: 2055.
- Kwon TJ, Kim TJ, Lee H, Min YW, Min BH, Lee JH, et al. Statin Use Decreases the Risk of Metachronous Gastric Cancer in Patients without Helicobacter pylori Infection. Cancers (Basel). 2021; 13: 1020.
- Seo SI, Park CH, Kim TJ, Bang CS, Kim JY, Lee KJ, et al. Aspirin, metformin, and statin use on the risk of gastric cancer: A nationwide population-based cohort study in Korea with systematic review and meta-analysis. Cancer Med. 2022; 11: 1217-1231.
- Su CH, Islam MM, Jia G, Wu CC. Statins and the Risk of Gastric Cancer: A Systematic Review and Meta-Analysis. J Clin Med. 2022; 11: 7180.
- Gonzalez FJ, Moschetta A. Potential role of the vitamin D receptor in control of cholesterol levels. Gastroenterology. 2014; 146: 899-902.
- Yin K, You Y, Swier V, Tang L, Radwan MM, Pandya AN, et al. Vitamin D Protects Against Atherosclerosis via Regulation of Cholesterol Efflux and Macrophage Polarization in Hypercholesterolemic Swine. Arterioscler Thromb Vasc Biol. 2015; 35: 2432-2442.
- Sun D, Cui X, Yang W, Wei M, Yan Z, Zhang M, et al. Simvastatin inhibits PD-L1 via ILF3 to induce ferroptosis in gastric cancer cells. Cell Death Dis. 2025; 16: 208.
- Sun D, Zhang M, Wei M, Wang Z, Qiao W, Liu P, et al. Ox-LDL-mediated ILF3 overexpression in gastric cancer progression by activating the PI3K/AKT/mTOR signaling pathway. Aging (Albany NY). 2022; 14: 3887-3909.








































































































































