Primary Mediastinal Embryonal Rhabdomyosarcoma in Adult: Literature Review and a Case Report
- 1. Department of Oncology Medicine, Comprehensive Oncology Center, México
- 2. Department of Internal Medicine, Medica Sur, México
ABSTRACT
Rhabdomyosarcoma is extremely rare in adults, but is the most common extra cranial solid tumor in children. Rhabdomyosarcoma in adults is a heterogeneous disease, with different form of presentation, histology, and prognosis and with difficulty in its treatment due to the rarity of the disease.
Here we briefly review the literature on adult rhabdomyosarcoma and present the case of a 39-year-old man clinical presentation of dyspnea, chest pain and weight loss, with initial approach with imaging studies and a mediastinal biopsy in which malignant neoplasia poorly differentiated with mesenchymal and epithelial immunophenotype is reported. He started chemotherapy with Paclitaxel/Carboplatin in a three-week regimen. After 2 cycles of chemotherapy a re-evaluation computed axial tomography showed progressive disease and the biopsy reported Embrionary Rhabdomyosarcoma with immunochemistry positive for myo D-1 and myogenine. Metastasis was confirmed at the diaphragm, retroperitoneal and central nervous system levels. 2nd line chemotherapy regimen with Vincristine, Doxorubicin and
cyclophosphamide were initiated.
KEYWORDS
Rhabdomyosarcoma;Chemotherapy;Embryonary ;Biopsy ; Mediastinal.
CITATION
Motola-Kuba D, Mackinney-Novelo I, Fernandez-Ferreira R (2018) Primary Mediastinal Embryonal Rhabdomyosarcoma in Adult: Literature Review and a Case Report. J Cancer Biol Res 6(1): 1111.
ABBREVIATIONS
RMS: Rhabdomyosarcoma; IRSG: Intergroup Rhabdomyosarcoma Study Group; CT or CAT: Computed Axial Tomography Scan; 18F-FDG PET/TC Scan: 2-deoxy-2- [fluorine-18]fluoro- D-glucose Integrated with Positron Emission Tomography /Computed Tomography; SUV: Standardized Uptake Value; ECOG: Eastern Cooperative Oncology Group; ERMS: Rhabdomyosarcoma Embryonal; ARMS: Rhabdomyosarcoma Alveolar; PRMS: Rhabdomyosarcoma Pleomorphic; SEER: Surveillance Epidemiology and End Results; NOS RMS: Not Otherwise Specified Rhabdomyosarcoma
INTRODUCTION
Rhabdomyosarcoma (RMS) comprises the most common soft tissue sarcoma in children and adolescents, accounting for more than 50% of soft tissue sarcomas in those age groups [1]. It is the third most common extra cranial solid tumor in children. On the other hand, RMS is extremely rare in adults; soft tissue sarcomas represent <1% of all adult solid tumor malignancies, and RMS accounts for only 3% of all soft tissue sarcomas in adults [2-4]. Intergroup Rhabdomyosarcoma Study Group (IRSG), formed on 1972 to investigate the therapy and biology of this disease mostly in children (by their definition, those younger than 21 years of age), has documented that increasing age is an adverse prognostic factor. The reasons for this age effect are unclear but may reflect the differences in histopathologic subtypes, primary anatomic origin, and clinical experience limited to case reports and small series. Resulting in worse long term survival ranging in 35-45% in adults, compared with children of 70-80% [5-7].
In this manuscript we provide a brief literature review on adult rhabdomyosarcoma and present a case report of a patient with the disease.
LITERATURE REVIEW
Rhabdomyosarcoma was first described by Weber in 1854, and classified as embryonal (ERMS), Botryoid, alveolar (ARMS), pleomorphic (PRMS). The histologic diagnosis of RMS has improved markedly with the current use of immunohistochemistry, electron microscopy and molecular genetic studies. Botryoid lesions are considered to be a variant of Embryonal RMS, with a better prognosis [8-10]. Later Mixed alveolar pleomorphic and not otherwise specified (NOS) were added to the histologic classification [10,11].
The histologic subtypes have different age distribution; the embryonal variant accounts for about 60% of all RMSs, and it has a bimodal age distribution, the large peak between 0 – 5 years and the second peak in adolescence [12-15]. Conversely ARMS is more common histologic subtype in adolescents. The percentage of the common pediatric subtypes (embryonal and alveolar histology) decline with age, whereas the pleomorphic subtype and the not otherwise specified Rhabdomyosarcoma are frequently histologic distribution in adult patients with RMS [16].
The exact cell of origin for RMS remains controversial, it has been suggested that multipotent mesenchymal stem cells can give rise to RMS; alternatively, other theories suggest that myogenic transformation can be induced in non-muscle cells by genetic manipulation and derangement of signaling pathways in different cells, resulting in de-differentiation and neoplastic transformation. The obscure origin of RMS is mainly due to the complexity of its presentation, the fact that it may originate at anatomic locations expected to be due of skeletal muscle, and variability among the histologic subtypes, different clinical behavior and prognosis. Suggesting that the different histologic subtypes may have different cellular origin [12,17].
RMS may arise from a wide variety of locations, and are grossly classified into extremity and axial lesions. Most frequent axial lesions are from head and neck, paraspinal region and genitourinary system. Abdominal and thoracic origin is rare in children and adolescents, although adults more frequently present with rare locations and worse prognosis [16].
Mediastinal Rhabdomyosarcoma is an extremely rare primary site of origin, as Intergroup Rhabdomyosarcoma Study Group (IRSG) defined, is an unfavorable site, with important vital structures nearby, difficult access for surgical resection, and that infiltration or displacement may compromise life. Most commonly mediastinum rhabdomyosarcoma occurs in association with germ cell tumor, teratoma or with thymic carcinosarcoma component. Primary mediastinal rhabdomyosarcomas unassociated with germ cells, teratomatous or malignant epithelial components are extremely rare, tend to have large size, important local invasion at diagnosis, and with aggressive behavior [4,18-20].
To the best of our knowledge there are 5 case report of primary mediastinal RMS unassociated with germ cell tumor, teratoma or carcinosarcoma. The case reported here does not have any component of germ cell, teratomatous or malignant or benign epithelial elements. This is one of the few cases of mediastinal rhabdomyosarcoma in adults, with uncommon embryonal histology presenting according to the age of the patient [11,21-23].
The differential diagnosis of mediastinal tumors with small blue cell tumors originating in the anterior mediastinum includes Lymphoblastic lymphoma, lymphocyte-rich thymoma, and small cell thymic carcinoma. Since the introduction of immunohistochemistry, the differential diagnosis is more accurate, with specific markers for epithelial, lymphoma, and rhabdomyosarcoma in addition to certain morphologic features that differentiates each other. The origin of these tumors in this location is uncertain; some experts have postulated that the sarcomatous elements may originate from myoid cells normally present in the thymus [23]. Although there is evidence of RMS originating in places devoided of myoid cells, and theories suggest tumors may arise from primitive, totipotential stem cells in the mediastinum.
The Intergroup Rhabdomyosarcoma Study Group (IRSG) was formed in 1972 with the goal of performing large randomized trials in children with RMS, to better describe the heterogeneity of the disease, determine the prognostic factors, improve multidisciplinary treatment and prognosis. Since the IRSG was formed, 4 trials have been designed, leading in a progress in the treatment of RMS and a 3 times increase of cure rates and benefit in overall survival, although most patients included in these trials were under 21 years old [24-27].
PROGNOSTIC FACTORS Y PROGNOSTIC GROUPS
One of the well-known prognostic factors in the pediatric group is histologic subtype, children with embryonal RMS had better prognosis with 5-year survival of 69% compared with 47% in alveolar RMS and 12% in pleomorphic RMS. The extension of the disease is another may or prognostic factor, patients with localized disease had better prognosis with 5-year survival of 82%, compared with regional disease 68% and distant disease 32%. The third known prognostic factor is the primary site of origin, head and neck and genitourinary (excluding bladder/ prostate) RMS with highest 5-year survival, and extremities, retro peritoneum, trunk, prostate, bladder and para-meningeal, with lowest survival, then head and neck and genitourinary are considered favorable sites with a 5-year survival of 5-year survival of 74% compared with unfavorable sites 53% [28].
Adults with rhabdomyosarcoma had significantly worse outcome than children with 5-year overall survival rates of 27% vs 61% respectively. Analysis of RMS cases registered in the Surveillance Epidemiology and End Results (SEER) public-access database has shown that a progressive decline in the survival curves with advancing age. Histologic subtype and tumor primary site are also important factors in adult patients according to multivariate analysis [16,28].
In accordance with the previously described risk factors, IRSG made a Pretreatment Staging Classification taking into account the primary site of origin, size of the lesion, Nodal infiltration and presence of metastasis; Stage 1: Orbit/eyelid, head and neck (excluding parameningeal), genitourinary (excluding bladder/prostate), all size tumor, without metastasis, regional nodes clinically involved by tumor or not; Stage 2 and 3: Bladder/ Prostate, extremity, parameningeal, other (trunk, retro peritoneum), ≤ 5 cm, without metastasis, however stage 3 includes regional nodes clinically involved by tumor and may be greater than 5cm; Stage 4 includes all sites of origin and sizes of tumor, without distant metastasis, and regional nodes clinically involved by tumor or not. The estimated 5- year survival by pretreatment stage for IRSG-III patients was as follows: Stage 1: 89%; Stage 2: 86%; Stage: 69%; and Stage 4: 30% [27,29]
Although clinic-pathologic characteristics of RMS are important for prognosis, postsurgical Pathologic results are also important for prognosis. The IRSG also grouped RMS according to completely resected tumors and localized disease (Group I), microscopic disease remaining (Group II), incomplete resection or biopsy with gross residual disease (Group III) and distant metastases present at onset (Group IV) [24-27].
Based on retrospective analysis of data from IRSG-III that identified patient subsets at different risks of failure, the IRSG defined risk categories, using pretreatment staging classification (Stage 1-4), and postsurgical residual tumor (Group I-IV). According to this, three risk groups were defined as follows: Low-risk patients were defined as those with primary tumors at favorable site (Stage 1, Group I-III) and unfavorable site (Stage 2, Group I); Intermediate-risk patients included favorable sites (Stage 1, Group II-III) and unfavorable anatomic sites (Stage 2-3, Group I-III); High risk patients were metastatic disease with any primary site (Stage 4, Group IV) [24-26,29,30].
TREATMENT
Despite the recent use of multimodality treatment protocols for RMS in children described by the Intergroup Rhabdomyosarcoma studies, have improved the overall survival, in adults the response rate and prognosis are uncertain [18,31- 33].
Some experts consider that childhood and adult RMS is a different disease, with different prognosis, and different response to treatments. There is no standard RMS treatment in adults. Short series had used multimodality approach, including wide resection of the primary tumor, radiation therapy for microscopic and macroscopic residual tumor, and multi agent chemotherapy. A wide variety of chemotherapeutic agents have shown activity in treating rhabdomyosarcoma including doxorubicin, vincristine, ifosfamide, cyclophosphamide and etoposide and actinomycin-D. However, determining the most effective combination has been complicated because of the rarity of the disease, poor information from case reports and series of cases, with a wide variety of treatment protocols [4,31,11].
CASE PRESENTATION
A 39-year-old male, previously healthy, with no medical co morbidities, none smoker, no alcohol or drug consumer presented in August 2015 with progressive shortness of breath, dry cough, thoracic pain and weight loss of 10 kg in the previous 3 months. On examination pale skin, rhythmic heart rate at 80 beats per minute, blood pressure 120/70 mmHg, Oxygen saturation with pulse oximeter at 88% on room air, without lymphadenopathy, with pleural effusion syndrome. Chest X-ray (posterior-anterior view) showed left pleural effusion and a mass in the left lung base. A computed axial tomography scan of the chest showed a multinodular mediastinal mass displacing the mediastinum, compresses the aorta, shifts the trachea and deforms the left bronchus. On September 2015 a TC-guided left pulmonary biopsy revealed malignant neoplasia poorly differentiated with mesenchymal and epithelial Immuno phenotype. A bone scan was negative and no other evidence of tumor in extension studies. He started chemotherapy on September 18th 2015 with Paclitaxel/ Carboplatin in a three-week regimen. After 2 chemotherapy cycles a re-evaluation computed axial tomography showed Progressive Disease.
He came to our center for a 2nd opinion in November 2015. The pathology biopsy was reviewed by the Pathology department, where they reported embrionary rhabdomyosarcoma with immunochemistry positive for myo D-1 and myogenine (Figure 1).
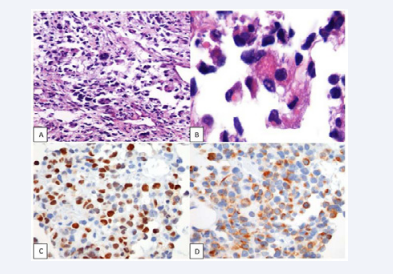
Figure 1 Hemtoxiline-eosin (HE) staining and immunohistochemical staining Figure 1A and Figure 1B: HE Staining of a retroperitoneal biopsy that shows an Embrionary Rhabdomyosarcoma. Figure 1C: Immunohistochemistry positive for myo D-1. Figure 1D: Immunohistochemistry positive for myogenine
In 18-F-FDG PET–TC scan a 15 cm hyper metabolic mass in the apical left hemi thorax that shifts the mediastinum and left lung, with SUV max 24.7, it extends to the diaphragm and involves it all. Hyper metabolism in right para-hilar lymphadenopathy, with SUV max 16.4, and retroperitoneal mass with no affection of abdominal structures of 3 cm in its anteroposterior diameter and SUV max 10 (Figure 2).
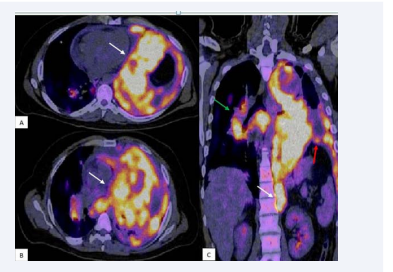
Figure 2 18F-FDG PET/CT scan Figure 2A and Figure 2B: In a transversal-section, a 15cm hyper metabolic mass in the apical left hemi thorax that shift the mediastinum and left lung, with SUV max 24.7 (white arrows). Figure 2C: RMS mediastinal that extends to the diaphragm and involves it all (red arrow). Hyper metabolism in right para-hilar lymphadenopathy (green arrow), and retroperitoneal mass with no affection of abdominal structures (white arrow)
Nuclear Magnetic Resonance reported 3 supratentorial millimeter nodules with contrast enhancement, and an infratentorial lesion in left cerebellar lobe (Figure 3).
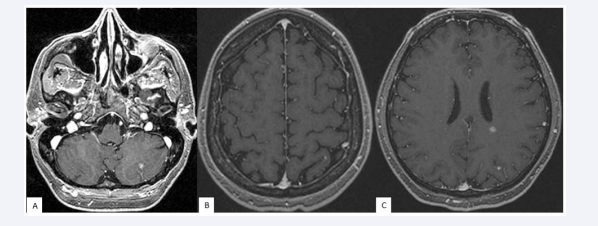
figure 3 Nuclear Magnetic Resonance. Figure 3A show a cerebellar lesion in the left cerebellar lobe (white arrow), Figure 3B and Figure 3C shows four supratentorial millimeter lesions with contrast enhancement (white arrows)
A port-a-cath was placed and 2nd line chemotherapy regimen with Vincristine, Doxorubicin and cyclophosphamide initiated in November 16th 2015. Gamma-knife of the cerebral lesion was realized. He received 3 chemotherapy cycles, and on January 2016, he was received on the Emergency department for oppressive chest pain, shortness of breath, a thorax CAT showed disease progression. He was in ECOG 3 and was put on Best Supportive Care; he died 1 month later.
CONCLUSION
Pure mediastinal embryonal rhabdomyosarcoma is extremely rare, with aggressive behavior, treatment extrapolated from children RMS has shown poor response rate and worse prognosis. There may have been more mediastinal RMS diagnosis, although might been misdiagnosed before immunohistochemistry use. It is important an accurate diagnosis, for multidisciplinary treatment and achievement of better treatments and outcomes for these patients.
The evolution of the patient presented here was concordant with the literature, with poor response to chemotherapy, symptoms associated with tumor infiltration to vital organs and poor prognosis.








































































































































